
Bonjour les copinous,
C’est noël et bien sûr on révise comme des foufous  … Ahah ah… ah… j’espère que ce n’est pas le cas pour tout le monde
… Ahah ah… ah… j’espère que ce n’est pas le cas pour tout le monde  … Bon du coups le nom dans le perfectionnement de la chimie organique j’ai un 'tit soucis contre intuitif que j’aimerais balayer avec l’appuis des experts :
… Bon du coups le nom dans le perfectionnement de la chimie organique j’ai un 'tit soucis contre intuitif que j’aimerais balayer avec l’appuis des experts :
Dans cette règle on fait intervenir les volumes les plus gros des orbitales avec les plus gros et les plus petit avec les plus petit.
En effet dans les réactions péricycliques on essaye d’expliquer la régioselectivité et la diastereoselectivité à l’aide des orbitales.
- Diastereoselectivité expliquée par la stabilisation d’un approche endo- plutot que exo- par exemple
- Regioselectivité expliquée avec ces notions de coefficients/volume des orbitales moléculaire
Dans cette dernière on doit étudier et prévoir le volume des orbitales selon les groupements attracteur ou donneur ramifiés sur le diène et le diènophile :
Par exemple on peut étudier la réaction suivante :
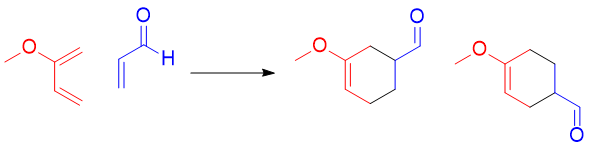
Alors on devra deviner lequel des deux est plus probable en s’essayant aux jeu de coefficient mis à disposition :
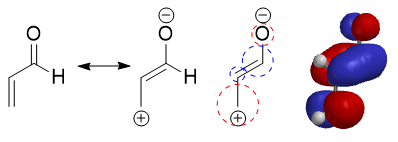
Et sur Spartan’16, cela confirmerais que sur une charge positive nous ayons plus de densité electronique. Autrement dit, une lacune electronique participe d’avantage à l’orbitale moléculaire que la charge négative ! Et là, dans ma tête ça débloque.
J’ai bien essayé de me dire, ok dans une LUMO on doit fonctionner à l’inverse que dans une HOMO et peut-être que ça a toujours un sens de parler de densité électronique avec un rapport à la mésomérie ? Et bien j’ai aussi eu de mauvaises expérience avec la simulation d’un benzaldéhyde où aucun site n’était franchement positif ni négatif…
Du coups j’aurais bien besoin d’un coups de main ! @Pierre_24 si tu passes dans le coin coin… 



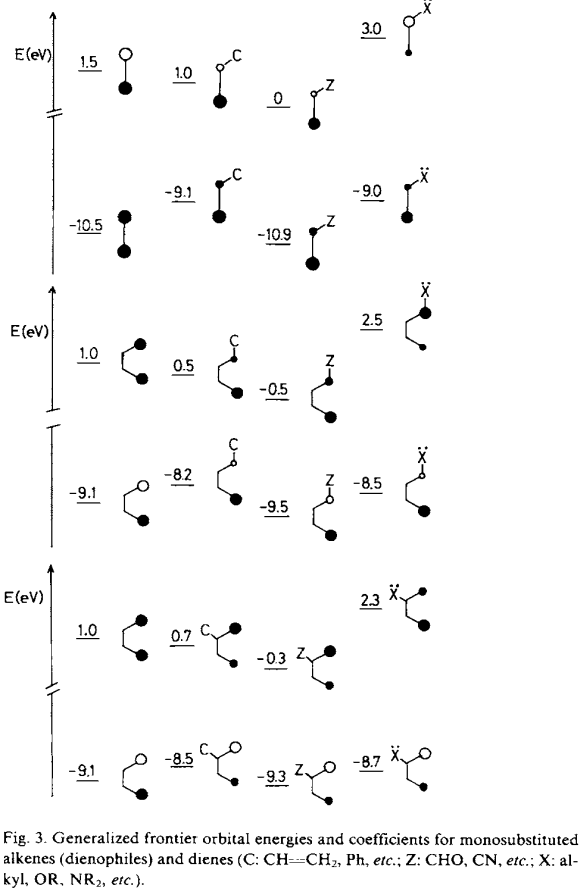

 Objectivement, tu ne peux pas. Le mieux que t’a sous la main pour le faire, c’est Hückel, et tu devrais normalement la tuner un peu pour intégrer des atomes qui ne sont pas des carbones. Je crois me souvenir qu’à un moment, on dit aussi fuck la FMO et qu’on regarde plutôt les formes mésomères, mais c’est un peu loin dans ma tête (je me souviens plus exactement du contexte).
Objectivement, tu ne peux pas. Le mieux que t’a sous la main pour le faire, c’est Hückel, et tu devrais normalement la tuner un peu pour intégrer des atomes qui ne sont pas des carbones. Je crois me souvenir qu’à un moment, on dit aussi fuck la FMO et qu’on regarde plutôt les formes mésomères, mais c’est un peu loin dans ma tête (je me souviens plus exactement du contexte).