
Bonjour à tous!
Voici un brouillon d'article sur la cheminformatics. Il n'est encore pas terminé et j'espère que vos commentaires m'aideront à l'améliorer.
N'hésitez pas à critiquer. 
Depuis les débuts de la chimie, des millions et des millions de molécules différentes ont été découvertes dans la nature ou synthétisées en laboratoire. La conservation et l'exploitation de cette masse de connaissance est un défi que la démocratisation de l'informatique nous permet enfin de relever efficacement. Le terme cheminformatics, mot anglais abbrégé de chemical informatics et généralement utilisé tel quel en français, peut se définir comme suit :
La cheminformatics consiste en la création, l'organisation, la gestion, l'extraction, l'analyse, la propagation, la visualisation et l'utilisation de données chimiques.1
L'objectif de cet article est de donner un aperçu modeste de cette discipline, de ses liens avec d'autres sciences et de son importance dans la recherche actuelle en chimie.
La représentation in silico des molécules
La question de la représentation des molécules est antérieure à l'apparition de l'informatique puisqu'il n'a jamais été possible de «voir» une molécule. Ainsi, au fil des siècles, les chimistes ont pris l'habitude de les dessiner avec toute une sémantique implicite. Désormais, à partir des représentations suivantes, tout chimiste est capable de calculer les masses molaires des molécules en question, d'estimer leur acidité, etc. :
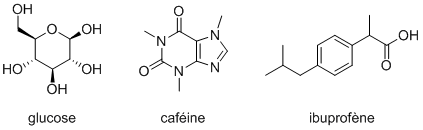
Malheureusement, un ordinateur n'est pas capable d'interpréter ces dessins tel que nous le faisons. Il a donc fallu inventer de nouvelles représentations adaptées aux ordinateurs. Deux grandes familles ont été développées : les notations inline (qui tiennent en une unique ligne) et les tables de données.
Une célèbre notation inline apparue dans les années 1980 et toujours très populaire aujourd'hui est la notation SMILES (Simplified Molecular Input Line Entry Specification).2 Celle-ci permet de décrire toute molécule sous forme d'une chaîne de caractère, ce qui permet de constituer des bases de données occupant peu de mémoire. Voici quelques exemples de SMILES :
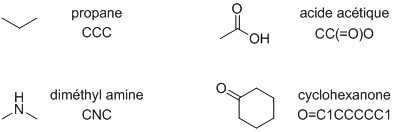
remplacer la diméthyl amine par l'éthanol!!!
Ensuite, il «suffit» d'écrire un parseur capable d'en déduire les atomes présents et les liaisons entre eux pour commencer à calculer certaines propriétés (masse molaire, etc.). Cette représentation a l'avantage d'être assez facilement écrivable et lisible par l'humain, mais elle souffre de quelques défauts. Le principal soucis est que l'on peut écrire plusieurs SMILES différents pour une même molécule (par exemple CCO ou OCC représentent toutes les deux l'éthanol). Des règles complexes ont donc été élaborées pour écrire des SMILES canoniques afin d'éviter d'enregistrer des doublons dans les bases de données.
L'autre format inline incontournable est le InChI (International Chemical Identifier).3 Il comble certaines lacunes des SMILES et fortement recommandé. En revanche, il est moins facilement interprétable ; j'en veux pour preuve le InChI de l'éthanol : 1S/C2H6O/c1-2-3/h3H,2H2,1H3.
Ces représentations inline ont l'avantage d'être légères mais ne peuvent pas contenir d'autres informations sur les molécules que leurs atomes et liaisons. Pourtant, il pourrait être intéressant d'associer à une molécule une géométrie donnée, c'est-à-dire les coordonnées de ses atomes en 2 ou 3 dimensions…
Les formats en tables répondent à cette demande. Les plus répandus sont le format XYZ et le molfile.4 Le premier contient le nombre d'atomes sur la première ligne, un commentaire à choix sur la deuxième puis une ligne par atome contenant son symbole atomique et ses coordonnées x, y et z. Pour une molécule d'eau avec une géométrie donnée, on pourrait avoir :
1 2 3 4 5 | 3 Eau O -0.58115 1.11467 0.02396 H 0.40818 1.12756 -0.01691 H -0.87051 1.39947 -0.87924 |
En revanche, il n'y a aucune information sur les liaisons entre les atomes et c'est au parseur de les établir en fonction des longueurs de liaisons, ce qui peut être ardu ! Le molfile, quant à lui, permet de gérer les liaisons avec sa table de connexion (j'ai volontairement simplifié la structure du fichier) :
1 2 3 4 5 | -0.5811 1.1147 0.0240 O 0.4082 1.1276 -0.0169 H -0.8705 1.3995 -0.8792 H 1 2 1 // l'atome 1 est connecté à l'atome 2 par une liaison d'ordre 1 1 3 1 // l'atome 1 est connecté à l'atome 3 par une liaison d'ordre 1 |
Pour résumer, d'un point de vue computationnel, moins de parsage est nécessaire pour extraire des informations des molécules avec les représentations en tables qu'avec les notations inline. Par contre, elles sont plus gourmandes en mémoire et moins intuitives à écrire pour les humains.
Bref, vous l'aurez compris, seule l'imagination est un frein à la création de nouvelles formes de représentations informatisées des molécules. À chacun donc de choisir le format qui convient en fonction de ce qu'il veut en faire.
Les éditeurs de molécules
Il n'est pas toujours facile d'écrire soi-même une molécule dans un format donné. C'est pourquoi en première ligne des programmes de cheminformatics, on trouve les éditeurs de molécules. Ce sont des programmes en ligne ou en version bureau qui permettent de dessiner rapidement n'importe quelle molécule puis de l'exporter sous toutes sortes de formats, y compris en images bien-sûr. Encore une fois, il existe une pléthore de ces éditeurs, à chacun de choisir le plus adapté à ses besoins.5 En voici deux, représentatifs de leur catégorie et open source :
L'éditeur web peut-être le plus connu est le JSME6, dans lequel je viens de dessiner l'aspirine. Vous pouvez l'essayer vous-même, il est minimaliste mais très intuitif (lien). Et pour la 3D, Avogadro est une référence.7
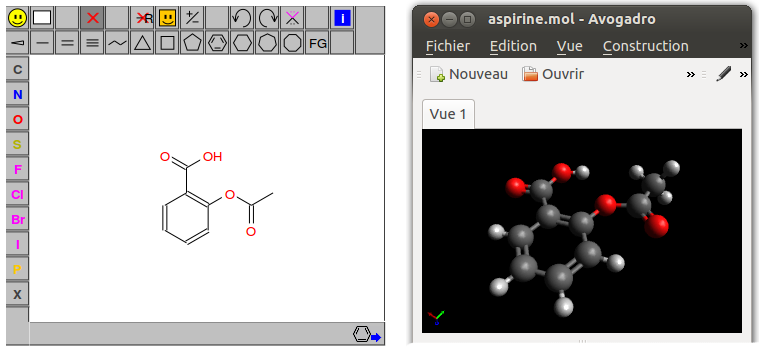
Avant de passer à la suite, citons encore l'excellent OpenBabel8, un outil complémentaire aux éditeurs capable de convertir entre eux plus de 110 formats de fichiers !
L'apport de la théorie des graphes
Au-delà des propriétés triviales à calculer telles que le nombre d'atomes d'azote présents ou la masse molaire, certaines requièrent plus d'efforts : la molécule contient-elle des cycles ? Quelle est la distance entre ces deux atomes ? Etc. Il n'est pas possible de répondre à ces questions directement à partir des représentations que nous venons de voir, c'est pourquoi ces représentations sont souvent converties en graphes en back-end des programmes de cheminformatics.
En mathématiques, un graphe est un ensemble de noeuds reliés entre eux par des arêtes. Il suffit alors de considérer les atomes d'une molécule comme des noeuds et ses liaisons comme des arêtes pour que l'on puisse appliquer la théorie des graphes aux molécules.
Illustrons tout ceci avec un exemple. Voici une molécule et son graphe :
FIG
À partir du graphe, on peut par exemple déterminer facilement le nombre de liaisons entre les atomes XXX et XXX en utilisant l'algorithme de Dijkstra (celui-ci permet de calculer le chemin le plus court entre deux noeuds).9
Comme vous pouvez le constater, la théorie des graphes est extrêmement puissante et nombreux sont ses théorèmes ou algorithmes utilisés pour résoudre des problèmes de chimie.
Les bases de données
Chaque jour, une quantité innombrable de nouvelles molécules sont publiées dans les revues de chimie du monde entier. Certaines organisations se sont données l'objectif de toutes les recenser dans des bases de données. Par exemple, le registre CAS a recensé et numéroté à ce jour plus de 89 millions de composés uniques !11 De nombreuses bases de données libres d'accès ou payantes sont sur le marché.12
Le principal intérêt des bases de données est d'y rechercher des molécules exactes ou des molécules contenant une sous-structure donnée. Par exemple, dans ChemSpider, si l'on dessine la sous-structure indole, on trouve entre autres les molécules suivantes :
FIG
Mais comment tout ceci fonctionne-t-il ? Encore une fois, les graphes sont utilisés ! Comparer la structure de deux molécules revient à rechercher des isomorphismes de leurs graphes.13
Applications
Jusqu'à présent, nous avons posé les bases de la cheminformatics : comment représenter les molécules in silico, les stocker, etc. Il est grand temps d'étudier ce qu'on peut faire de tout ceci: QSAR, … .
Cheminformatics != chimie computationnelle
Une dernière précision avant la fin : il ne faut pas confondre la cheminformatics et la chimie computationnelle, bien que ces deux disciplines sont basées sur l'informatique et qu'elles se recoupent souvent. Nous l'avons vu, la cheminformatics consiste avant tout à manipuler des molécules enregistrées au format informatique et leurs données associées. La chimie computationnelle, elle, modélise des molécules. Dans ce cas, la puissance des ordinateurs est utilisée pour faire de gros calculs de chimie quantique et calculer toutes sortes de propriétés telles que les énergies des électrons, la forme des orbitales, la structure d'intermédiaires réactifs dans des réactions, etc.
Toute recherche appliquée en chimie computationnelle utilise des outils de cheminformatics, puisqu'en quelque sorte ils constituent l'interface humain-machine, mais l'inverse n'est pas vrai.
En apprendre plus sur la cheminformatics
Cet article s'est efforcé de donner un aperçu des problèmes principaux rencontrés en cheminformatics et des liens avec les autres sciences, telles que les mathématique ou la pharmacologie. Si vous souhaitez suivre la recherche actuelle en cheminformatics, deux revues spécialisées existent (toutes deux en anglais bien-sûr) :
- Journal of Cheminformatics (libre d'accès)
- Journal of Chemical Information and Modeling (accès payant)
- Molecular Informatics (accès payant)
Autrement, de nombreux blogs plus accessibles sont régulièrement mis à jour par des personnes clés du milieu :
- Blog de Marcus D. Hanwell (l'un des développeurs de Avogardo)
- Blog de Noel O'Boyle (l'un des développeurs de OpenBabel)
- Blog de Tim Vandermeersch (l'un des développeurs de OpenBabel et auteur de Helium)
- Blog de Prof. Henri S. Rzepa (Imperial College London) (l'un des auteurs du format CML)
Conclusion
Nous voici arrivés à la fin de cet article. Je serai ravi de répondre à toutes vos questions sur le sujet et à discuter de ce domaine passionnant plus en profondeur dans les forums du site.
Merci pour la lecture.
-
Engel, Thomas. J. Chem. Inf. Model. 2006, 46, 2267–2277 (lien, consulté le 20 juillet 2014) ↩
-
Weininger, David. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36 (lien, consulté le 20 juillet 2014), SMILES - A Simplified Chemical Language (lien, consulté le 20 juillet 2014), OpenSMILES specification (lien, consulté le 20 juillet 2014) ↩
-
Heller, Stephen. McNaught, Alan. Stein, Stephen. Tchekhovskoi, Dmitrii. Pletnev, Igor. Journal of Cheminformatics 2013, 5, 7 (lien, consulté le 26 juillet 2014) ↩
-
Dalby, Arthur. Nourse, James G.. Hounshell, W. Douglas. Gushurst, Ann K. I.. Grier, David L., Leland, Burton A.. Laufer, John. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255 (lien, consulté le 27 juillet 2014) ↩
-
ChemDraw, la référence en milieux académique et industriel (propriétaire, payant) (lien, consulté le 20 juillet 2014), ChemSketch, une bonne alternative mais uniquement sur Windows (propriétaire, gratuit) (lien, consulté le 20 juillet 2014), MarvinSketch, une bonne alternative multi-plateformes (propriétaire, gratuit) (lien, consulté le 20 juillet 2014), BKChem, multi-plateformes et open-source mais développement stoppé (libre, gratuit) (lien, consulté le 20 juillet 2014), JChemPaint, également multi-plateformes et open-source et développement stoppé (lien, consulté le 20 juillet 2014), ChemDoodle, excellent éditeur online (libre, gratuit) (lien, consulté le 20 juillet 2014) ↩
-
Bienfait, Bruno. Ertl, Peter. Journal of Cheminformatics 2013, 5, 24 (lien, consulté le 20 juillet 2014) ↩
-
Avogadro - Free cross-platform molecule editor (lien, consulté le 20 juillet 2014) ↩
-
OpenBabel : The Open Source Chemistry Toolbox (lien, consulté le 20 juillet 2014) ↩
-
Le pathfinding avec Dijkstra (lien, consulté le 2 août 2014) ↩
-
Zamora, Antonio. J. Chem. Inf. Comput. Sci. 1976, 16, 40–43 ↩
-
ChemSpider (libre d'accès) (lien, consulté le 20 juillet 2014), PubChem (libre d'accès) (lien, consulté le 20 juillet 2014), The Cambridge Crystallographic Data Centre (libre d'accès) (lien, consulté le 20 juillet 2014), SciFinder (accès payant) (lien, consulté le 20 juillet 2014), Reaxys (accès payant) (lien, consulté le 20 juillet 2014) ↩
-
Isomorphisme de graphe (lien, consulté le 2 août 2014) ↩



 ) à la chime quantique, ce qui est un choix, mais je trouverai intéressant de mentionner en deux mots les simulation de type monte-carlo/dynamique moléculaire.
) à la chime quantique, ce qui est un choix, mais je trouverai intéressant de mentionner en deux mots les simulation de type monte-carlo/dynamique moléculaire.