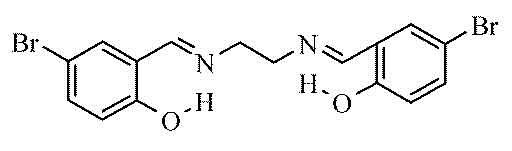
Je dois analyser un spectre, normalement il correspond à cette espèce :
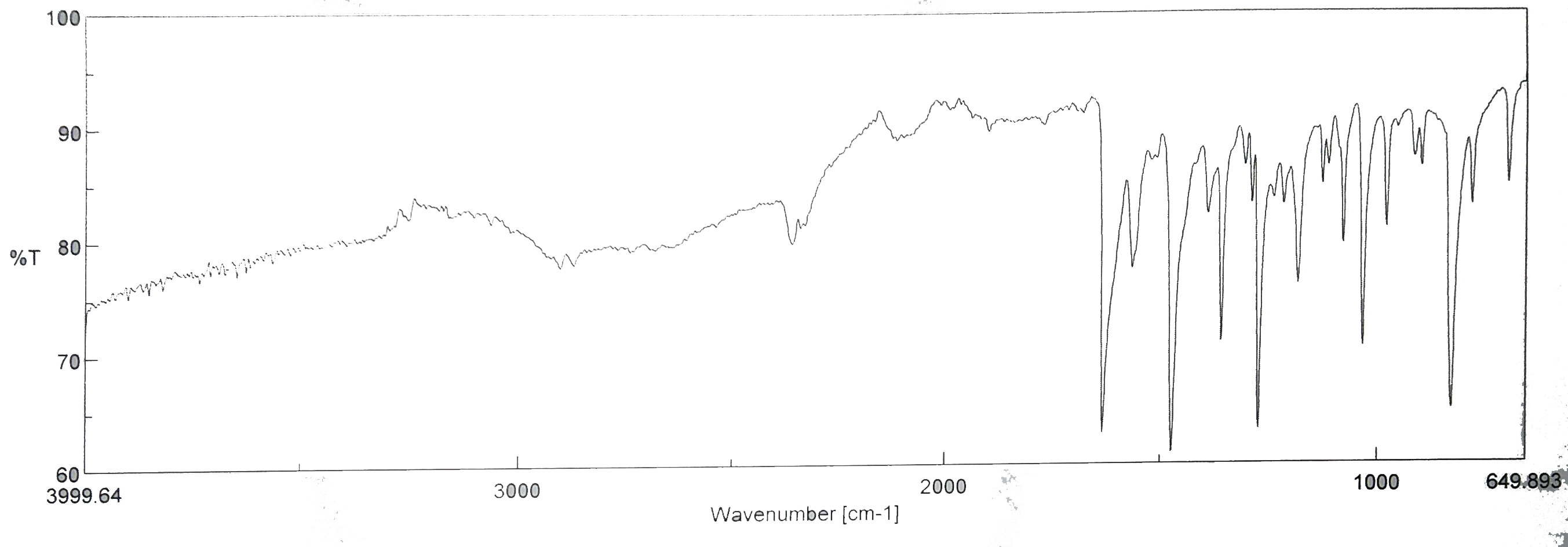
J’ai donc obtenu ce spectre là, et l’on me dit de ne pas identifier les pics inferieur à 1000 1/cm
Spectre Infra-Rouge
j’ai pu identifier ces pics là :
Pic 2360: CO2
Pic 1635: C=N
Pic 1466: C=C(arom)
Mais je ne sais pas si c’est suffisant ? On m’a dit de ne pas chercher à faire dire au spectre, ce que j’ai besoin qu’il dise. En gros je dois chercher quelques pics très "caractéristiques" mais je n’y connais pas grand chose.
J’suis plutot du genre à chercher ce dont j’ai besoin, voir "si ça colle" et hop x).
Si quelqu’un pouvait m’aiguiller sur "comment tout identifier". Merci de votre lecture et aide future
Salut! Bon, d'abord, tu sais sûrement que les pics inférieurs à 1000 1/cm font partie de la zone fingerprint qui n'est pas facile à exploiter. Je ne pense pas que tu trouveras beaucoup d'informations utiles dans cette zone. Entre 1000 et 2000 1/cm, tu as des pics dus à la déformation angulaire et 2000 à 4000 1/cm, des pics d'élongation. D'après moi, si tu réussis à analyse la plupart des pics principaux (tous les C-H, O-H, C=C, C=O, C=N, etc.), cela suffit, parce que le reste peut être dû à du bruit de fond ou des impuretés. D'ailleurs, avant de vérifier si ça colle, tu devrais simplement essayer d'identifier les bandes caractéristiques sans penser à ta molécule (des protocoles d'identification sont faciles à trouver sur le Web) et seulement après vérifier si ton spectre correspond à ta molécule. Sinon, tu risques de ne pas être impartial.
De toute façon l'IR c'est pour confirmer si ta synthèse a réussi et que tu as bien retrouvé tes liaisons chimiques. Tu dois avoir une table des différents groupements chimiques avec la valeur associée et l'allure du spectre (grande-moyenne-fine dépression, etc)… Tu le dis toi-même, il faut voir si c'est compatible avec ce que tu sais de ta molécule.
Tu dis que tu as identifié CO2, le CO2 gaz carbonique ou j'ai mal compris ta notation ? Parce que sinon c'est étrange.
Et il faut faire attention au fait que plusieurs groupements vont être associés aux mêmes valeurs de bande.
Tu as identifié un pic de liaison C=N, de groupemennt aromatique; u dois pouvoir trouver des pics correspondant aux liaisons C-OH, voire C-Br.
De façon générale quand tu récupères un spectre tu connais la formule brute (spectro de masse par exemple), et tu essaies d'inférer la structure de la molécule à partir des infos du spectre. Donc chaque pic doit être analysé et dire avec quoi il est compatible, là tu dois en avoir une dizaine à analyser. Il t'en manques donc, c'est le principe de l'analyse par IR / par RMN.
Ah tiens zone fingerprint ? Je ne connaissais pas ce terme, c'est à cause des traces de graisses que nous laissons ?
Il est impératif d'identifier si ces pics proviennent d'élongation ou de déformations ?
Je vais voir au niveau des protocoles d'identification, ça m'as l'air d'être pile ce que je cherche !
PS : D'après ma RMN, et toutes les caractéristiques que j'ai relevé, c'est la bonne molécule à 100% sure donc j'aurais eu le droit d'être impartial, mais j'aimerais apprendre la vraie méthode.
EDIT : Goeland-croquant
Ouais du CO2 gazeux, car on respire à coté de l'appareil, d'ailleurs pour placer la fine poudre sur le diamant c'est très compliqué, alors on a le nez dessus et on respire (histoire de vivre)…
Toute la bande de gauche (>3000 1/cm) me parait bizarre, mais toutes les personnes ayant synthétisé dans le labo cette molécule on eu cette espèce de patate.
Dans la zone avant 1000 cm-1, c'est une zone de signature de ta molécule (et elle lui est propre), donc si ta molécule existe dans une base de données tu la compares et tu regardes. C'est le boulot d'un ordi si la molécule est déjà connues, sinon tu travailles avec les déplacements entre 1000 et 3000 cm -1 en général.
Écoutez le chimiste l'autre biologiste, moi je ne suis qu'un biologiste qui a fait un cours d'orga.
Non, la zone fingerprint s'appelle ainsi parce qu'il y a plein de pics très caractéristiques et donc très durs à identifier, comme si c'était les empreintes digitales de la molécule. C'est presque impossible à exploiter par un être humain.
Les pics d'élongation d'un groupement sont différents des pics de déformation pour le même groupement. Mais bon si tu as tu as un protocole, osef un peu. D'habitude, je commence toujours par vérifier s'il y a une énorme bande entre 3500 et 3200 1/cm, c'est O-H et tu ne peux pas le manquer. Après ça il y a plusieurs bandes C-H qui différent par leur type de vibration à 2962, 2872, 1450, 1375 1/cm… L'alcool est aussi assez visible à 1260-1000 1/cm. Pour C=C, c'est différent si la molécule est linéaire ou aromatique. Mais il existe des listes qui disent le type de vibration, le nombre d'onde, et surtout l'intensité qui est assez pratique à connaître.
Sinon, en vrai, maintenant les ordis récupèrent tout et analysent directement le spectre. (La seule question c'est est-ce que le labo a le logiciel pour, mais voilà pour la théorie. Avoir une table de références est impératif.
Bah moi je suis chimiste, hein quand même. Mais pas trop habitué aux analyses, surtout quand il s'agit de faire ce qu'un prof demande… Si vous voyez ce que je veux dire.
J'ai enormement de tables dans mes cours, bien heureusement.
Personnellement je découpe mon spectromètre en 6 zones d'analyses : 5 fonctionnelles entre 4000 et 1500 cm-1 et une spécifique entre 1500 et 700 cm-1. Et je sors ma trousse de crayons de couleur, je trace des trait pour séparer les zones, j'écris au dessous ce que je devrait y voire puis j'essaye de mettre des couleurs sur tout les pics correspondant au même type de groupe que j'identifie.
La première zone de 4000 à 3000 cm-1, je cherche la "patate" caractéristique des alcools ou ou des amides et la bande fine des alcyne qui se répète pour confirmer vers 2100 cm-1. On peut aussi avoir des pics qui vont nous dire si nous sommes en présence d'une amine primaire, secondaire ou tertiaire ce qu'on pourras également trouver plus en détail dans la dernière zone.
La deuxième zone, juste en dessous de 3000cm-1 à 2500cm-1, c'est la zone des vibration C-H sur un carbone sp3 et au C-H des aldéhydes donc en général si c'est plat c'est qu'il n'y en a pas, sinon c'est le bordel ici. On peut aussi y trouver la vibration du O-H de COOH, c'est aussi une "patate" très caractéristique, il faudrait que tu voit quelques exemples.
ensuite il se passe pas grand chose d’intéressant jusqu'à 2200, la zone des triples liaison CC et CN, c'est là qu'on vient confirmer la bande fine qu'on a trouvé dans la première zone.
La zone 4, de 2000 à 1600 cm-1 on y voit des C=O mais c'est plus ou moins la seule indication, un tas de groupement apparaissent ici : esters, acides, aldéhydes, cétones …
Zone 5 ! de 1600 à 1500 cm-1, c'est là qu'on trouve les c=c des alcènes et des aromatiques.
La dernière zone en dessous de 1500 cm-1 jusqu'à 700 cm-1 est assez compliquée à analyser, c'est la zone des vibration C-C, C-O et C-N. Cette zone est pratiquement unique à chaque molécule, un ordinateur et une grosse base de donnée est indispensable.
En gros, l'IR tout seul ça va rarement te permettre de trouver la formule développé d'une molécule et dans ce cas là ça me parait compliqué.
Ce spectre est un peut moyen en plus :/ t'as rien avant 1700cm-1 et la ligne de base est super basse. Tu l'as fait comment ?
edit :
la fine poudre sur le diamant c'est très compliqué
Bah ça explique pourquoi c'est moche… Nous avions de quoi pastillé sur KBr et foutre le tout dans une autre machine à IR. Mais les profs ne voulaient pas… Alors on s'est un peu demerdé comme on a pu. ça serait cool d'avoir plus d'explication Akio, sur le pourquoi on doit pas utilisé de poudre par ATR ?
J'ai fais une correction d'ailleurs… Spécifique à l'ATR si j'me trompe pas. Parce qu'un spectre Diamant c'est pas un Spectre "normalisé". (j'suis le seul à avoir pris cette initiative, si on pouvait me dire si c'était une bonne idée xD)
Tout simplement parce que c'est pas fait pour ça il existe des accessoire pour l'ATR des poudres, mais en général c'est assez cher et les applications sont assez spécifiques (produits trop absorbants pour la transmission "classique", impossibilité de broyer ou faire fondre …).
Si encore tu avais ce genre d'accessoires à disposition ça serait bon mais vu que tu parles de diamant, on est sur de l'ATR horizontale sur une surface très (trop) petite (un petit Perkin a interféromètre de Michelson tout beau tout propre ? j'aime bien ces appareils).
De plus l'intensité de l’absorption est dépendante de la longueur d'onde en ATR.
Le meilleur moyen d'obtenir un bon spectre sans artefacts c'est clairement la transmission, tant que le produit est compatible avec le nujol/pastilles de KBR/fenêtres de CaF2/NaCl… après c'est sure, ça demande plus d'entrainement que l'ATR pour faire de bonnes pastilles.
C'est vrai mais c'est un peu dommage, c'est l'une des machines que nous avons déjà utilisé en TP (celle qui fonctionne aux pastilles de KBr). Là on était dans un cycle de TP un peu particulier, d'où la nécessité d’interpréter les choses un peu plus profondément.
Connectez-vous pour pouvoir poster un message.
Connexion
Pas encore membre ?
Créez un compte en une minute pour profiter pleinement de toutes les fonctionnalités de Zeste de Savoir. Ici, tout est gratuit et sans publicité.
Créer un compte



 donc j'aurais eu le droit d'être impartial, mais j'aimerais apprendre la vraie méthode.
donc j'aurais eu le droit d'être impartial, mais j'aimerais apprendre la vraie méthode.
 il existe des accessoire pour l'ATR des poudres, mais en général c'est assez cher et les applications sont assez spécifiques (produits trop absorbants pour la transmission "classique", impossibilité de broyer ou faire fondre …).
il existe des accessoire pour l'ATR des poudres, mais en général c'est assez cher et les applications sont assez spécifiques (produits trop absorbants pour la transmission "classique", impossibilité de broyer ou faire fondre …).