
TW: noob en théorie des graphes
Salut,
En tant que chimiste théoricien, j’ai l’occasion de mesurer des paramètres géométriques tels que les angles dans des molécules. Par exemple,
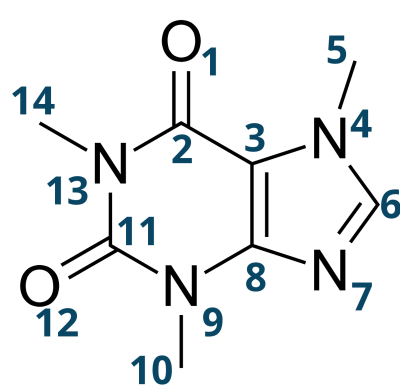
dans cette molécule, on voudrait par exemple connaitre l’angle entre les atomes 2, 3 et 4. La question qui se pose alors est "comment lister tout les angles possibles de cette molécule?".
Bien entendu, il s’agit d’un problème de théorie des graphes. Dit autrement, je cherche un algorithme qui me permet de lister tout les sous-graphes connecté de k-sommets (k=3 pour les angles, mais je suis également intéressé par k=4) dans un graphe non-dirigé (l’angle entre les atomes 2, 3 et 4 est évidement le même que celui entre les atomes 4, 3 et 2), mais qui peut contenir des cycles, comme le montre l’exemple ci-dessus.
Bien entendu, un "bête" algorithme bruteforce consisterai à lister tout les k-uplet de sommet, et à vérifier qu’ils sont effectivement connectés. Sauf que vu que le graphe est pas directionnel, il faut également que je supprime les doublons.
Est ce qu’il y aurai un alogorithme un peu plus malin ? Je me doute bien que ça va être un algoritme à la complexité algorithmique dégueulasse (O(nk) ?) mais peu m’importe, tant que je suis sur de les avoir tous, et une seule fois 
Merci d’avance 
EDIT: lorsqu’on fait des recherches sur Google, on est très vite orienté sur le problème des cliques. Or ce n’est pas ce que je veux, puisque je ne recherche pas des sous-graphes complets 


 Mais ça ressemble un peu dans l’esprit à ce qui était dit juste au-dessus. Tu peux peut-être construire tes solutions récursivement.
Mais ça ressemble un peu dans l’esprit à ce qui était dit juste au-dessus. Tu peux peut-être construire tes solutions récursivement. Grillé par Aabu !
Grillé par Aabu !



