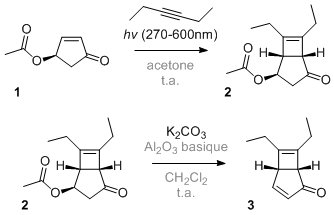
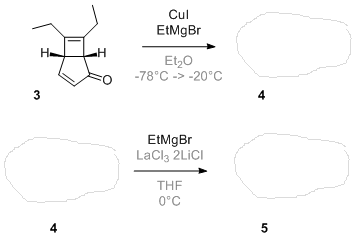
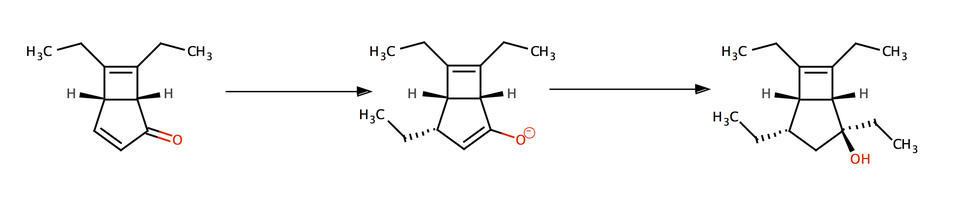
Bonjour à tous,
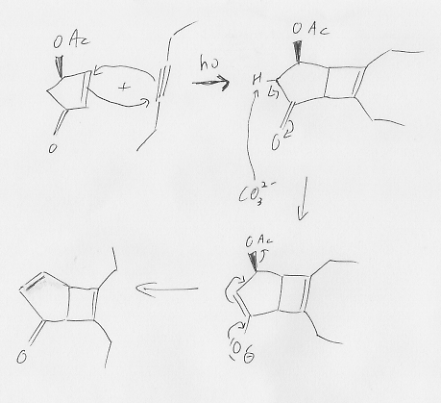
Je suis tombé hier sur un article reportant une très jolie synthèse totale d'une molécule naturelle. Je vous propose d'en faire un exercice car la plupart des étapes sont des réactions bien connues. Le but est de compléter les trous et je donnerai au fur et à mesure la suite de la synthèse. Détaillez la stéréochimie autant que possible, c'est intéressant et toujours instructif! Je ne donne volontairement pas le nom de la molécule ni la référence de l'article.  (je le ferai à la fin pour éviter tout soucis de copyright)
(je le ferai à la fin pour éviter tout soucis de copyright)
C'est parti:
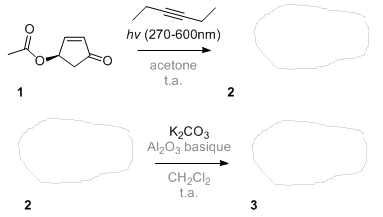
J'ai mis en gris les conditions réactionnelles que vous pouvez "ignorer".
Si vous n'avez pas d'éditeur de molécules sous la main, je vous recommande d'utiliser http://web.chemdoodle.com/demos/sketcher et de faire des captures d'écran.
N'hésitez pas à vous lancer!

 [/HS]
[/HS]



 , mais ayant vue sur un modèle moléculaire des tensions angulaire des bases opioïdes récemment, la regle de Bredt me parait un peu trop empirique.
, mais ayant vue sur un modèle moléculaire des tensions angulaire des bases opioïdes récemment, la regle de Bredt me parait un peu trop empirique. )
)