Ce que tu as trouvé sur internet, c'est pour mettre un atome interstitiel dans du Ti FCC, sans le déformer. Du coup, on peut calculer $a$ (le paramètre de maille) à partir du rayon du Ti. Dans ton cas, c'est un peu différent, puisqu'on a un alliage. Les atomes de Ti ne se touchent pas forcément, mais c'est pas grave, puisque le paramètre de maille est donné. Au passage, "se touche", c'est pas correct, juste une simplification.
Du coup, dans le lien que tu as donné, seules les relations avec a sont utiles
$$
\frac{a}{2} \geq R(Ti) + R(Octa)
$$
$$
\frac{a\sqrt{3}}{4} \geq R(Ti)+R(Tetra)
$$
De ces équations, tu peux trouver les rayons des différents sites. Avec les données que tu donnes, tu trouves que les sites octahédriques sont suffisamment spacieux pour accueillir n'importe lequel de tes deux atomes, et le sites tétrahédriques sont trop petits. Ce que ça dit, c'est que tu vas en effet avoir une déformation, en plus de l'agrandissement du paramètre de maille.
Maintenant, concernant l'inversion, c'est une grande question. Déjà, ça changerait la stoichiométrie de ton alliage, et il faudrait vérifier que tu peux avoir un tel alliage de manière stable. Ensuite, ça augmenterait les déformations, ce qui semble rédhibitoire, mais qui ne serait pas non plus exceptionnel (mais c'est probablement ce qui est demandé ici). Il y a de nombreux alliages de fer avec des atomes d'yttrium, qui sont pourtant énormes. Il faut aussi réaliser que même si ce n'est pas stable, si la température est suffisamment basse, le réagencement des atomes sera extrêmement lent.
En réalité, les rayons atomiques ne peuvent que donner une idée très génerale du résultat. Ça va dépendre beaucoup du comportement des électrons (orientation des orbitales, hybridisation, …). Simuler le système avec un code de théorie de la fonctionelle de la densité te donnerait un résultat bien plus précis.
Pour la dernière question, qui est indépendente de la précédente, il faut utiliser le ŕesultat de la troisième question et les données de l'exercice. Si tu as des difficultés pour cette question, on peut aussi en parler rapidement.

 :
:

 ) ou que tu espères qu'un autre membre voit quelque chose que j'ai manqué.
) ou que tu espères qu'un autre membre voit quelque chose que j'ai manqué.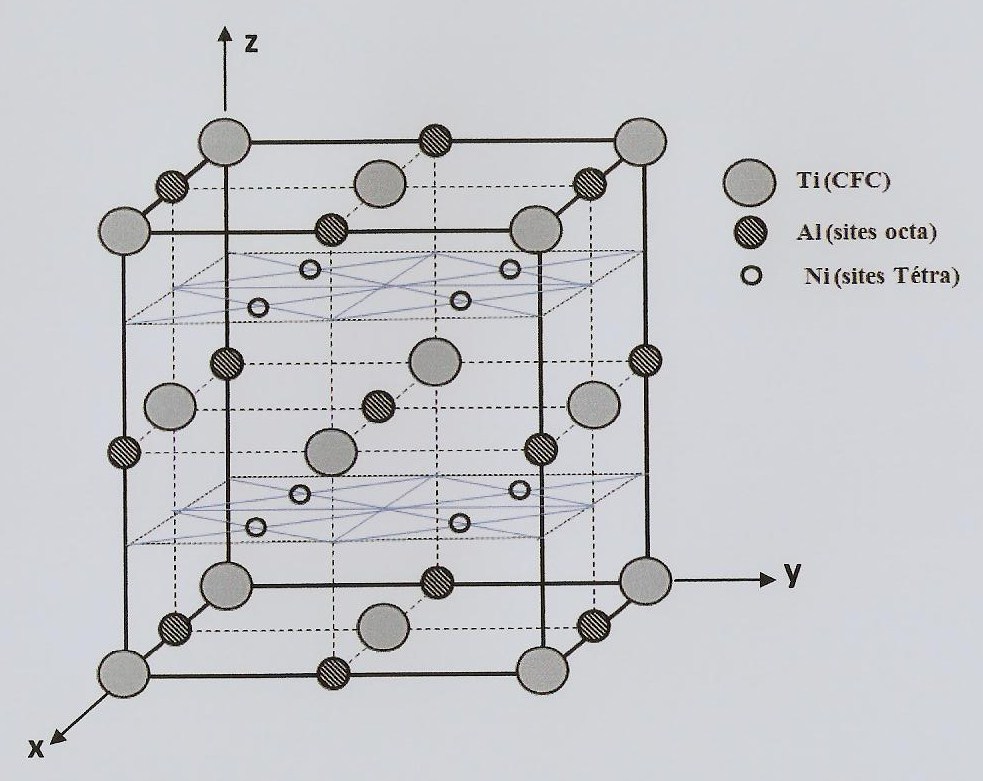{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}