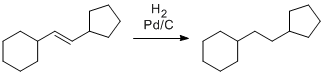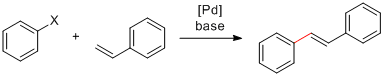Je souhaiterais d'ici quelques jours publier en bêta un article de chimie (il en faut aussi!) que je suis en train d'écrire. Il ne me reste qu'à ajouter quelques schémas pour qu'il soit compréhensible. Quel est le meilleur moyen de le faire? Un copier-coller dans ce topic?
Voilà, il ne manque plus qu'une photo et un schéma à ajouter puis la grosse relecture générale et l'ajout des dates de consultation des liens en référence. Je suis conscient de m'être attaqué à un sujet extrêmement vaste, donc oui, je pourrais encore ajouter 1000 choses… Soyez cléments sur ce point, mais sinon allez-y sans retenue avec les critiques!
Pour expliquer mon approche, voici comment est né cet article. Je me suis dit qu'il serait intéressant d'en écrire un qui soit:
accessible (je l'espère) aux débutants ayant quelques notions de chimie organique
avec un peu de culture générale autour (qu'est-ce que la catalyse, l'historique, le Nobel, …)
pas trop académique (avec des exemples réels d'applications)
qui aborde les dernières avancées de la recherche
Et à ma connaissance, il n'y a rien de tel sur le web à l'heure actuelle…
La synthèse artificielle de molécules a pris un rôle important dans notre société au cours du XXème siècle. Est-ce un bienfait? On peut en discuter sur le forum. Quoi qu'il en soit, la plupart de ces molécules (médicaments, parfums, colorants et autres) sont des composés organiques, c'est-à-dire des molécules constituées principalement d'atomes de carbone reliés entre eux. Voici quelques exemples:
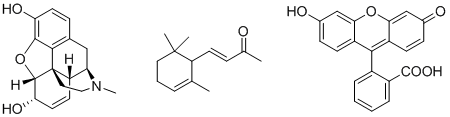
Morphine | $\alpha$-Ionone | Fluorescéine
La morphine1 est un anti-douleur naturel bien connu, l'$\alpha$-ionone2 est l'un des composants du parfum naturel de violette et la fluorescéine3 est une molécule artificielle qui émet de la lumière sous ultraviolets. Comme nous pouvons le constater, la structure de ces molécules repose bel et bien sur un squelette d'atomes de carbone. Le développement de toute une palette de réactions chimiques capables de former ces liaisons C-C est donc très utile en chimie organique.
2) Les réactions catalytiques
Les couplages croisés catalysés au palladium sont des réactions dites catalytiques car elles fonctionnent grâce à un catalyseur. Un catalyseur est une espèce ajoutée dans le milieu réactionnel destiné à accélérer ou rendre possible une réaction chimique sans être consommé ni intégré au(x) produit(s). Parfois, le catalyseur peut même être récupéré à la fin de la réaction et réutilisé. Les réactions catalytiques sont classées en deux grandes familles: la catalyse hétérogène et la catalyse homogène.
La catalyse hétérogène
Dans ce type de catalyse, le catalyseur et le(s) réactif(s) ne sont pas dans la même phase (liquide, gaz, etc.). C'est par exemple le cas lorsque le catalyseur est un solide et que les réactifs sont des gaz, ou lorsqu'un catalyseur solide est mis en suspension dans une solution contenant les réactifs.
Un bon exemple de la vie de tous les jours est le pot catalytique4 qui équipe les voitures modernes. Il s'agit d'un filtre constitué d'un alliage métallique à travers lequel les gaz d'échappement passent. Au contact de l'alliage, plusieurs réactions chimiques sont accélérées, dont la réaction ci-dessous. L'objectif du pot catalytique est de convertir un maximum de CO en CO2 car le CO2 est moins nocif que le CO:
$2CO+O_2\rightarrow 2CO_2$
Pot catalytique
Un autre exemple est l'hydrogénation d'alcènes avec du palladium sur charbon Pd/C. Ce mélange de palladium et de graphite est une poudre noire plus ou moins fine et non soluble que l'on ajoute dans une solution contenant l'alcène. Une pression d'hydrogène gazeux (H2) est ensuite appliquée sur la solution et l'alcène se fait hydrogéner en alcane:
Hydrogénation avec Pd/C
La catalyse homogène
L'autre grande famille est la catalyse homogène, où le catalyseur et les réactifs sont dans la même phase. La plupart du temps, ils sont tous les deux dissous en solution.
Les couplages croisés catalysés par le palladium font partie de cette famille, comme nous allons le voir plus loin. Mais prenons déjà un autre exemple plus simple enseigné dès les premiers cours de chimie organique: l'estérification. Cette réaction bien connue est le mélange d'un acide carboxylique avec un alcool. La réaction peut fonctionner sans catalyseur, mais lorsqu'on ajoute un peu d'acide dans le milieu (H2SO4, HCl, etc.), elle est accélérée:
Estérification en milieu acide
3) Les couplages croisés
Une réaction de couplage croisé permet de former une liaison entre deux fragments de molécules grâce à la présence d'un catalyseur métallique. Ces deux fragments ne réagiraient pas en l'absence du catalyseur. Si ces deux morceaux sont identiques on parle d'homocouplage, et s'ils sont différents de couplage croisé. À l'heure actuelle, il est possible de former toutes sortes de liaisons: C(sp2)-C(sp2), C(sp2)-N, C(sp)-(sp2), C(sp3)-C(sp3), etc. Cet article se focalisera sur les liaisons C(sp2)-C(sp2) puisque historiquement ce sont les premières à avoir été développées:
Liaisons C(sp2)-C(sp2)
Avant les années 1970, très peu de méthodes existaient pour former de telles liaisons directement. Puis sont arrivés les travaux de Kumada et Corriu5. Dès lors, l'intérêt pour les couplages croisés n'a cessé de grandir et des centaines d'articles scientifiques sont publiés chaque année sur le sujet, repoussant toujours les limites de ces réactions passionnantes.
Les ingrédients
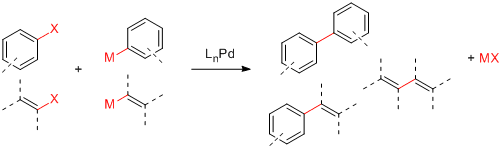
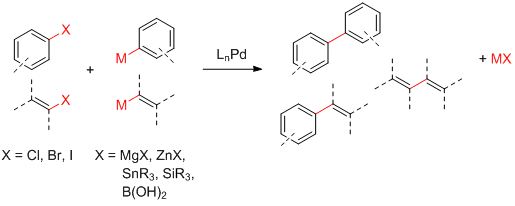
Après tous ces beaux discours, passons au vif du sujet. Les trois ingrédients de base dans un couplage croisé traditionnel sont:
un réactif halogéné (R-X)
un réactif organométallique (R'-M)
un catalyseur au palladium (LnPd)
Voici quelques combinaisons possibles:
Ingrédients pour un couplage croisé
Les couplages croisés ont été développés pour une grande variété de réactifs organométalliques différents. À l'origine, Kumada a exploité les réactifs de Grignard (R'-MgX). Ceux-ci sont simples à préparer mais ne tolèrent pas la présence de carbonyles à cause de leur réactivité bien connue avec ces derniers.6 Negishi7 a obtenu de meilleurs rendements pour les produits biaryles avec le zinc (R'-ZnX2). Stille8 et Hiyama9 ont quant à eux développé de nouveaux réactifs basés sur l'étain (R'-SnR3) et le silicium (R'-SiR3). Finalement l'un des meilleurs couplages est le fameux couplage de Suzuki10 qui utilise des acides boroniques ou des dérivés (R'-B(OH)2, R'-B(OR)2, etc.).
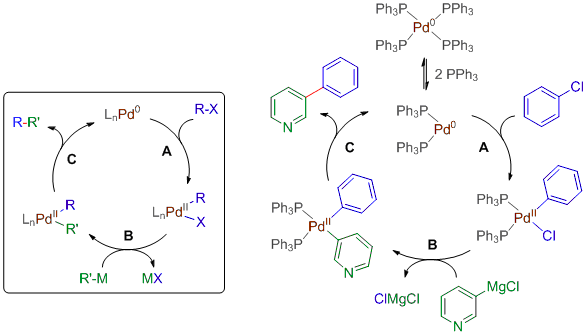
Le cycle catalytique
Les réactions citées ci-dessus suivent toutes la même séquence d'étapes élémentaires suivantes à quelques détails près: addition oxydante, transmétallation et élimination réductrice. Ces étapes constituent un cycle: la dernière étape régénère le système, lui permettant ainsi de répéter la séquence tant qu'il y a des réactifs à consommer. Voyons tout ceci en détail en étudiant le cycle catalytique général pour tout couplage croisé ainsi qu'un exemple particulier de couplage de Kumada catalysé par le complexe de palladium Pd(PPh3)4:
Cycle catalytique général | Exemple de couplage de Kumada
1) La première étape est l'addition oxydante (A): le composé halogéné s'additionne sur le palladium et fait passer son état d'oxydation de 0 à II. D'un point de vue mécanistique, on peut dire que Pd0 s'insère dans la liaison C-X du composé halogéné. Remarquons que dans notre exemple de couplage de Kumada, deux ligands PPh3 doivent préalablement se dissocier du complexe Pd0(PPh3)4 pour générer le catalyseur actif Pd0(PPh3)2.
2) La deuxième étape est la transmétallation (B): le groupe R' du composé organométallique R'M est transféré d'un métal à l'autre (de Mg à Pd dans le cycle de Kumada) et prend la place de l'halogène sur le palladium. L'halogène qui se trouvait sur le palladium est quant à lui éliminé avec le métal du réactif organométallique.
3) La dernière étape est l'élimination réductrice (C): les deux groupes R et R' sont éliminés pour former le produit de la réaction tout en régénérant le catalyseur. L'élimination est dite réductrice car le palladium passe de II à 0.
Conformément à la définition d'une réaction catalytique, le catalyseur est régénéré à la fin de la réaction. Ainsi, pour 1 mole de réactifs, il n'est pas nécessaire de charger 1 mole de catalyseur: 0.1 mole ou moins peuvent suffire. Certaines réactions industrielles tournent même avec 0.00001 mole de catalyseur par mole de réactifs, c'est-à-dire que chaque molécule de catalyseur fait 100'000 cycles au cours de la réaction!
Ces trois étapes constituent la base de toutes les réactions de couplages croisés mais de petites variations peuvent exister. La réaction de Suzuki requiert par exemple une base tandis que la réaction de Hiyama nécessite une source de fluorure. Je vous encourage à jeter un coup d'œil à ces autres cycles catalytiques dans un livre ou sur internet, vous verrez que le principe est toujours le même.
La course à la performance
Au cours de ces 40 dernières années, beaucoup de recherches ont été menées pour mieux comprendre chaque étape et ainsi optimiser la réaction. Premièrement, l'efficacité de l'addition oxydante dépend de l'halogène: l'insertion du palladium 0 dans une liaison C-I est beaucoup plus facile que dans une liaison C-Cl, tout simplement car l'énergie de liaison est plus faible:
Liaison
Énergie de liaison
C-I
238 kJ/mol
C-Br
285 kJ/mol
C-Cl
330 kJ/mol
C-F
485 kJ/mol
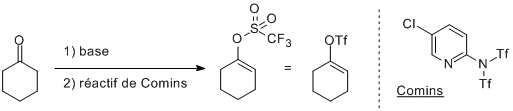
Comme alternative aux halogènes, il est possible d'utiliser les triflates C-OTf. Ceux-ci sont assez facile à préparer et sont très réactifs (ils se situent entre les C-I et les C-Br). Plusieurs réactions permettent de les faire, en voici juste un exemple:
Préparation d'un triflate
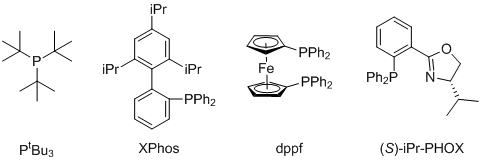
Le choix de l'halogène est important, mais l'axe d'optimisation le plus important est sans aucun doute la modification des ligands du catalyseur. Au-delà de la simple triphénylphosphine PPh3 que nous avons vue, des centaines d'autres phosphines sont désormais dans le commerce: liste de phosphines. Impressionnant n'est-ce pas?
Plusieurs tendances ont été constatées. Premièrement plus les phosphines sont riches en électrons plus l'addition oxydante se fait facilement (ex: PtBu3). C'est logique: le palladium est mieux armé (plus nucléophile) pour s'insérer dans la liaison C-X. Pour accélérer l'élimination réductrice, les "gros" ligands (encombrés stériquement) sont meilleurs. L'encombrement favorise l'élimination du produit pour que les ligands se sentent à nouveau plus libres autour du palladium. Il existe ensuite les ligands bidentés (plus stables, ex: dppf), les ligands chiraux (pour les réactions énantiosélectives, ex: (S)-iPr-PHOX), etc. etc. Parmi les ligands couramment utilisés on rencontre le XPhos car il cumule plusieurs de propriétés mentionnées ci-dessus.
Ligands courants
Comment s'y retrouver parmi tous ces ligands? En général, lorsqu'on veut faire un "simple" couplage, on commence par tenter la réaction avec un catalyseur classique tel que le Pd(PPh3)4 ou le Pd(dppf)Cl2. Si ça ne fonctionne pas, on cherche alors quelque chose de plus sophistiqué en fonction de ce qu'on a obtenu comme produits/sous-produits.
4) De la théorie à la pratique
Avant de continuer, passons à un chapitre un peu plus récréatif: des applications réelles de la réaction de Suzuki. La première provient de mon cursus tandis que la seconde a été publiée par une entreprise pharmaceutique dans le cadre du développement d'un médicament potentiel contre certaines maladies pulmonaires.
Expérience personnelle
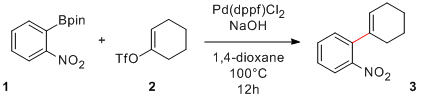
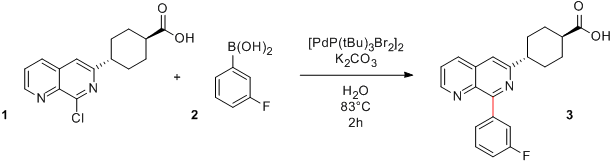
Voici donc un couplage de Suzuki effectué pour préparer le nitrobenzène substitué 3, un composé dont j'avais besoin. Il existe certainement d'autres conditions réactionnelles ou méthodes pour synthétiser cette molécule, mais la réaction employée dans le cas présent s'est avérée efficace et a fourni le produit avec un bon rendement de 91%:
Couplage de Suzuki
Quels sont les points importants à prendre en compte pour concevoir le montage? Il y en a principalement trois:
La température à atteindre est de 100°C. Le point d'ébullition du dioxane étant de 101°C, il faut utiliser un réfrigérant et effectuer la réaction à reflux pour éviter que le solvant ne s'évapore.
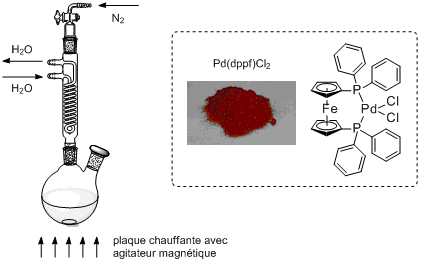
La réaction est sensible à l'oxygène (O2). En effet, l'oxygène peut oxyder le catalyseur actif Pd0 en PdII, ce qui le désactiverait. Il faut donc effectuer la réaction sous atmosphère inerte. Pour ce faire, j'ai connecté le haut du réfrigérant à une ligne de Schlenk,11 ce qui permet de faire le vide dans le système pour aspirer tout l'oxygène puis d'y remettre de l'azote (N2).
Pour ajouter les réactifs sans devoir retirer le réfrigérant, l'idéal est d'utiliser un ballon bicol muni d'un septum. Une seringue peut transpercer le septum pour ajouter les liquides et le septum peut être retiré sous contre-flux d'azote pour ajouter les solides.
photo du montage
Une fois la verrerie assemblée, il ne reste plus qu'à lancer la réaction. Le catalyseur (37 mg, 5 mol%) et l'ester boronique (249 mg, 1 mmol), tous deux des solides, sont chargés dans le ballon "à sec" puis le montage est mis sous atmosphère inerte. Du dioxane dégazé (15 mL) puis une solution aqueuse de NaOH 3 M dégazée (3 mL, 1 mmol) sont ensuite ajoutés. Finalement, le triflate (276 mg, 1.2 mmol) est ajouté et le tout est chauffé à 100°C pendant une nuit. Après extraction, concentration et purification par chromatographie sur colonne, le produit final est obtenu avec 91% de rendement (184 mg, 0.91 mmol).
Comme vous pouvez le constater, il n'y a rien d'insurmontable. Ce couplage a bien fonctionné dès le premier essai, mais ce n'est pas toujours le cas: il faut parfois varier plusieurs paramètres avant de rencontrer le succès. Tout est question d'expérience (et de chance!).
Application industrielle
Ces réactions ne sont pas uniquement utiles en milieu académique sur quelques dizaines de milligrammes, elles sont également largement exploitées dans l'industrie de la chimie fine. En 2011, un article qui recense des dizaines d'exemples de couplages croisés utilisés à grande échelle a été publié dans la revue Chemical Reviews.12 Comme promis, en voici un qui a servi pour obtenir un candidat potentiel contre certaines maladies pulmonaires:13
Couplage de Suzuki
La réaction a été menée avec 58.2 g de 1 et 33.6 g de 2 en chargeant seulement 0.4 mol% de catalyseur et a fourni 60 g de produit, soit un très bon rendement de 88%. Il est intéressant de noter que le couplage s'est produit entre l'acide boronique de 2 et le chlore de 1 et non entre l'acide boronique de 1 et le fluor de 1, ce qui démontre en partie que l'addition oxydante se fait préférentiellement avec les halogénures dont l'énergie de liaison C-X est la plus faible (cf. tableau du chapitre 3)!
5) Quelques réactions dérivées
En parallèle aux réactions que nous avons vues jusqu'ici, qui permettaient essentiellement de former des liaisons C(sp2)-C(sp2), d'autres réactions similaires ont été développées.
Historiquement la réaction de Heck a été l'une des premières réactions de couplage croisé. Elle forme également une liaison C(sp2)-C(sp2), mais en se passant du réactif organométallique. Elle permet le couplage d'un réactif halogéné avec une double liaison. Elle est donc extrêmement utile en synthèse totale puisqu'on peut alors facilement attacher des fragments sur toute molécule possédant une double liaison. Le cycle catalytique est quelque peu différent, je vous laisse le consulter vous-même:14
Couplage de Heck
Passons à un autre type de liaison: C(sp2)-C(sp). Il s'agit du couplage de Sonogashira qui permet d'attacher un alcyne terminal sur un carbone hybridé sp2. Dans cette réaction, un co-catalyseur de cuivre est utilisé pour former du réactif organométallique cuivré in situ à partir de l'alcyne terminal au fur et à mesure de l'avancement de la réaction:15
Couplage de Sonogashira
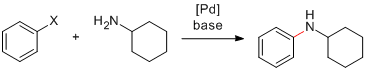
Finalement, une réaction synthétiquement intéressante est celle de Hartwig-Buchwald puisqu'elle forme une liaison C-N entre un aryle et une amine primaire ou secondaire. En général les ligands bidentés ou encombrés stériquement sont à privilégier pour éviter la formation de sous-produits non-désirés. 16
Couplage de Hartwig-Buchwald
6) Les activations C-H
Les activations C-H sont des réactions très récentes et prometteuses. Un réactif possédant une simple liaison C-H peut y être utilisé à la place du réactif halogéné! On utilise le terme activation car la liaison C-H en question est considérée comme intrinsèquement non-réactive et doit être activée. C'est par exemple le cas des liaisons C-H du benzène ou du pentane. En revanche, une liaison C-H en position $\alpha$ d'une cétone est considérée comme réactive puisqu'on peut facilement la casser par déprotonation et n'entre donc pas en considération dans ce chapitre.
Pour comprendre ce que signifie activation, rappelons ce qu'est une réaction bimoléculaire en solution: deux réactifs qui nagent dans un solvant et entrent en collision aléatoirement. Toutes ces collisions n'aboutissent pas forcément à une réaction, il faut que la collision soit efficace (avec suffisamment d'énergie et selon la bonne orientation des groupes fonctionnels). Pour des réactions "simples" tel qu'un transfert de proton, presque toutes les collisions seront efficaces. En revanche, entre un catalyseur au palladium et une liaison C-H d'un benzène, il faudra peut-être des milliards de collisions avant que le palladium ne s'insère dans la liaison (addition oxydante). En d'autres termes, la réaction ne fonctionnera pas.
En activation C-H, puisque les collisions entre le catalyseur et la liaison C-H ne sont pas assez souvent efficaces, la stratégie consiste à trouver un moyen de diriger le palladium vers la liaison et le forcer à s'y insérer. Pour ce faire, il faut que le réactif portant la liaison C-H porte aussi un groupe directeur.
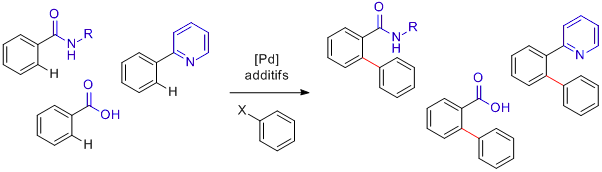
Prenons comme exemple l'arylation C-H en position ortho. Plusieurs groupes fonctionnels peuvent servir de groupes directeurs. Parmi les plus simples on trouve les amides, les acides carboxyliques ou les 2-pyridyles:
Activations C-H avec groupes directeurs
Le cycle catalytique peut différer d'une réaction à l'autre, mais voici une proposition générale communément admise:17
cycle catalytique
L'une des différences avec le cycle étudié auparavant est l'état d'oxydation du palladium, qui varie entre II et IV au lieu de 0 et II. Mais concentrons-nous surtout sur le rôle du groupe directeur. Grâce à celui-ci, le catalyseur PdII(OAc)2 peut activer et rompre l'une des liaisons C-H en position ortho du groupe directeur. Ensuite, une addition oxydante suivie d'une élimination réductrice délivre le produit. Finalement, pour régénérer le catalyseur, de l'acétate d'argent est utilisé: les ions Ag+ ayant une forte affinité avec les halogénures, ils arrachent facilement ces derniers du palladium.
Ces activations C-H sont donc très attractives lorsque le produit que l'on souhaite synthétiser porte déjà un groupe utilisable comme groupe directeur car celui-ci permet l'économie d'une liaison C-X. Et ceci n'est que le début de l'ère des activations C-H! Pour vous en convaincre, vous pouvez jeter un coup d'œil aux publications du groupe de recherche de Jin-Quan Yu: activation CH en position meta, activation de liaisons C(sp3)-H, réactions énantiosélectives, etc.!
7) Le Prix Nobel de chimie 2010
Le développement de toute cette chimie méritait bien un Prix Nobel. Ce fût chose faite en 2010 lorsque la récompense fût attribuée conjointement à Richard F. Heck, Ei-ichi Negishi et Akira Suzuki, soit trois grands chimistes parmi les pionniers du domaine:18
"pour les couplages croisés catalysés par le palladium en synthèse organique"
8) Conclusion
Apparus dans les années 1970, les couplages croisés catalysés par le palladium sont devenus des réactions centrales en chimie organique et sont désormais massivement utilisés tant en milieu académique qu'en industrie. À l'heure actuelle, des milliers de groupes de recherche à travers le monde n'ont cesse de repousser les limites de ces réactions passionnantes.
Cet article n'est qu'une modeste introduction à tout ce pan de la chimie et avait pour objectif de présenter les concepts de base puis de donner envie d'en apprendre plus. De nombreux thèmes n'ont donc pas pu être abordés. La principale omission est certainement que le palladium n'est plus l'unique métal de transition exploitable: des couplages croisés avec du nickel, du rhodium, du ruthénium ou même du fer ou du cobalt ont été développés entre-temps. Ces deux derniers sont particulièrement intéressants en terme de coût puisque ce sont des métaux bon marché.
Bref, il y aurait encore beaucoup à dire, mais arrêtons-nous là! N'hésitez pas à commenter et à venir poser toutes vos questions sur le forum Sciences.
En fait, je suis exactement ton public cible, chimiste avec le background qui commence à devenir intéressant, et du coup, même si je suis un peu fâché avec la chimie orga, je peux lire et comprendre ton texte. Ce qui signifie qu'il a atteint son but, et me concernant, c'est une victoire en soit. Ça veut dire aussi que c'est relativement bien écrit, que les trucs se suivent logiquement et qu'on a envie de continuer. Bon, par contre, tu prend le parti de t'adresser à des personnes qui ont le niveau pour le comprendre, parce qu'il y a énormément de termes non-défini ! (ce qui ne me choque pas, mais pourrait rebuter le tout venant). J'ai aussi cette facilité, parce que c'est écrit comme un article scientifique, avec ces codes, cette habitude de citer tout ce qui passe, cette manière de présenter des choses (c'est limite si tu met pas des numéros à toutes tes molécules: « alors, 23 réagit avec 49 et 61 pour donner le produit 7b … » ), bref, c'est bien quoi
Côté "technique", comme j'ai pu le mentionner, je suis en froid avec la chimie orga, je ne peut donc pas vraiment t'aider de ce côté là. Rien ne m'as en tout cas "choqué" quand j'ai lu le texte, ce qui veux tout et rien dire. J'ai fait appel à Blackline qui devrait à mon avis avoir plus de trucs à raconter de ce coté là. Bien entendu entendre que "le palladium est relativement bon marché" a toujours le don de me faire ouvrir grand les yeux, mais à force, on s'y fait (et c'est vrai que comparé à certaines terres rares, c'est très (!) bon marché).
Bon, par contre, tu prend le parti de t'adresser à des personnes qui ont le niveau pour le comprendre, parce qu'il y a énormément de termes non-défini ! (ce qui ne me choque pas, mais pourrait rebuter le tout venant).
Je plussoie. Il serait préférable de définir certaines notions pour Monsieur Lambda en note de bas de page. Le reste, AMHA, ça passe bien.
Edit : par contre, arrêtez avec les titres introduction/conclusion. C'est un article, pas un mémoire. Une phrase de transition et hop, ça passe crème.
Bon, par contre, tu prend le parti de t'adresser à des personnes qui ont le niveau pour le comprendre, parce qu'il y a énormément de termes non-défini ! (ce qui ne me choque pas, mais pourrait rebuter le tout venant).
En fait c'est vrai que quand je dis "accessible aux débutants", c'est peut-être un peu exagéré. C'est dur à estimer.
(c'est limite si tu met pas des numéros à toutes tes molécules: « alors, 23 réagit avec 49 et 61 pour donner le produit 7b … » ), bref, c'est bien quoi
Honnêtement je trouve que ces numéros font un peu trop "technique" et je pensais plutôt les enlever.
Bien entendu entendre que "le palladium est relativement bon marché" a toujours le don de me faire ouvrir grand les yeux, mais à force, on s'y fait
Je n'ai pas lu en détail (mais je compte bien le faire lorsque l'article sera publié, vues les critiques positives que tu reçois ici), par contre je ne suis pas d'accord sur le format des références bibliographiques : il me semble qu'il n'y a pas de raison, ici, pour qu'elles soient données sous la forme de numéro en exposant avec renvoi à la fin. Je pense qu'il serait bien plus adapté d'utiliser des liens directement dans le texte (qui correspondent mieux à ce qu'on a l'habitude d'avoir sur le web).
J'ai regardé quelques articles publiés précédemment, et il n'y a pas de consensus clair sur la façon d'organiser les références - d'ailleurs ton article est probablement l'un des plus soignés de ce point de vue. Cependant, il n'y a vraiment aucune raison de produire cette longue bibliographie à la fin, qui ne donne aucune information supplémentaire (par rapport aux bibliographies dont on a l'habitude dans les documents scientifiques). Des liens directs seraient moins surprenants, auraient un meilleur rendu, et seraient plus pratiques (y compris, je suppose, pour les logiciels qui analysent le texte, comme des robots ou des logiciels pour personnes non-voyantes).
Voilà. Si tu trouves que c'est du pinaillage, tu as parfaitement raison, et je n'insisterai pas.
J'ai regardé quelques articles publiés précédemment, et il n'y a pas de consensus clair sur la façon d'organiser les références - d'ailleurs ton article est probablement l'un des plus soignés de ce point de vue. Cependant, il n'y a vraiment aucune raison de produire cette longue bibliographie à la fin, qui ne donne aucune information supplémentaire (par rapport aux bibliographies dont on a l'habitude dans les documents scientifiques). Des liens directs seraient moins surprenants, auraient un meilleur rendu, et seraient plus pratiques (y compris, je suppose, pour les logiciels qui analysent le texte, comme des robots ou des logiciels pour personnes non-voyantes).
En fait, mathiasm applique "bêtement" (ce n'est pas un reproche) ce qui se fait dans le monde scientifique en matière de citation de référence (ça et l'habitude de citer des noms de personnes). L'avantage, c'est que toutes les références sont à la fin de l'article, parce que c'est ce qui se fait d'habitude. Ceci étant dit, j'avoue que c'est pas forcément adapté à ce qui se fait ici (c'est pratique sur une version papier ou tu peux mettre les feuilles les unes à coté des autres, moins sur le web)
Comme le dit pierre_24, je ne me suis pas posé trop de questions. Tous ces liens n'ont pas vocation à être consultés en direct lors de la lecture de l'article donc dans cette optique ce n'est pas grave si ce n'est pas très pratique de devoir descendre au bas de la page (d'après moi). L'objectif premier est de donner au lecteur la possibilité de vérifier lui-même ce que je raconte si besoin (comme dans un article scientifique, et toute autre discipline j'espère). Ensuite le deuxième objectif est de fournir un point de départ pour les lecteurs qui veulent aller plus loin, mais encore une fois je ne pense pas qu'ils le feront à la première lecture en direct…
Par contre là où j'ai mis les liens "en dur" comme le propose katana, c'est quand je veux que le lecteur les consulte en direct. Il y en a deux (la liste des phosphine et la page de Yu).
Si je mettais tous les autres liens de cette manière, j'aurais peur que le lecteur se sente obligé de cliquer dessus pour continuer et s'y perde… Peut-être qu'un compromis serait bien, par exemple ne pas devoir descendre au bas de la page:
Très interessant comme article ! Alors après une première lecture attentive je relèverais quelque points :
Les couplages croisés ont finalement pour but d'avoir plusieurs réactifs capables de réagirs entre eux et ainsi donner plusieurs molécules à la fin ou c'est autre chose ? Car dans l'article ce n'est pas précisé… (alors que le mot catalyseur y est defini…). Car on y voit dans l'une de tes premières équation, plein de réactif donner plein de produit.
Pourquoi montrer un derivé halogené qui réagit avec une amine à l'aide de Pd sans en montrer le mécanisme ? En soit dès les premières année en chimie organique on sait qur R-X et les amines réagissent, j'imagine que le rendement doit en être bien amélioré mais, un petit bout de mécanisme supplémentaire ne ferait pas de mal ?
Même remarque pour Sonogashira… Une base déproton l'alcyne ok, du coups il devient bon nucléophile, le rôle du métal est-il si important ? Je pense que oui vue que c'est ce que l'article essaye de mettre en avant
(Alors soit, les réactions que je cite sont en fait "pas terrible" et nécessite IMPLICITEMENT ces catalyseur, au quel cas je m'en excuse ^^. soit tu as oublié de mettre en avant que cela augmenter considérablement le rendement ou une petite illustration pour marquer le coups)
Anyway, c'est un super article et je te félicite pour la prise d'initiative !
Si tu parles bien du schéma "Ingrédients pour un couplage croisé", je n'avais pas pensé qu'il pouvait porter à confusion, merci de le relever! Non non le but des couplages croisés n'est pas de faire un gros mélange tel que dessiné, le but est de coupler un réactif halogéné avec un réactif organométallique. Là j'ai juste donné toutes les combinaisons possibles avec une seule flèche pour avoir un rendu visuel plus cool qu'en écrivant quatre équations. (Je constate à l'instant qu'il y a une erreur de typo sur le schéma, faudra que je corrige)
Concernant Hartwig-Buchwald, je ne souhaite pas détailler le mécanisme (c'est un cycle similaire à celui détaillé plus haut et il est donné en référence). Ça commencerait à faire beaucoup de cycles dans l'article et le rendrait trop scolaire! J'ai écrit ce chapitre "5) Quelques réactions dérivées" juste pour montrer qu'on pouvait faire d'autres couplages au palladium que des C(sp2)-C(sp2). En revanche, je vais rajouter quelques phrases à propos de la "simple" attaque directe de l'amine sur l'aromatique puisque justement ça ne marche bien que dans de rares cas et que Hartwig-Buchwald résout le problème. Merci pour l'idée!
Pareil, pas trop de cycles. Par contre je ne crois pas qu'un alcyne déprotoné puisse attaquer un aromatique halogéné… (à vérifier, mais de tête je n'ai jamais vu ça)
Anyway, c'est un super article et je te félicite pour la prise d'initiative !
Je viens de me lancer dans un autre: l'hydrogénation catalytique. Mais pas d'emballement, j'ai commencé celui sur le palladium en novembre-décembre je crois. Il a mis beaucoup de temps à mûrir.
Tout couple carbanion/électrophile n'est pas forcément apte à réagir: il faut que le premier soit assez nucléophile et que le second assez électrophile (et d'autres facteurs). Je vais voir si je trouve un exemple dans la littérature chimique.
Concernant une SNAr entre une amine et cycle aromatique halogéné, je n'ai trouvé que ça: Efficient Nucleophilic Aromatic Substitution between Aryl Nitrofluorides and Alkynes. Mais ça ne fonctionne qu'avec un NO2 en position ortho et aucun groupe électro-donneur ailleurs sur le cycle… Et ils précisent en introduction que très peu de méthodes existent en dehors du couplage de Sonogashira. Toutes ces méthodes alternatives sont exotiques et limitées (micro-ondes, InCl3, AgI + PPh3, etc).
Mais bien sur ! Que suis-je CON. Putain c'est ma semaine à dire des conneries… Tu as tout juste à ce sujet. Les SNAr (ouais parce que j'ai pas tilté que c'était aromatique, j'ai consideré pour tout R) se font avec des desactivants. On préférera tout de même des sulfonations si tu veux garder le coté "Réversible" et récupérer ton benzène.
Merci pour la prise en consideration de ma question nulle ! purée t'es bien patient
Je relance le sujet 2 mois après, désolé j'ai été extrêmement occupé!
Maintenant ma grande question c'est: article ou tutoriel? Il y a quelques semaines, dans un sujet du forum que je ne retrouve malheureusement plus, beaucoup de gens critiquaient les articles qui prenaient plus de 10 min à lire. D'un autre côté, ce n'est pas vraiment un tutoriel dans le sens où il ne donne qu'un aperçu du domaine… Qu'en pensez-vous?
Sinon il ne reste qu'un schéma à dessiner et go validation!
Bonne nouvelle, j'ai "terminé"! Plus qu'à changer une ou deux couleurs dans les schémas pour rester consistent du début à la fin. Si vous avez des remarques, c'est le dernier moment avant que j'envoie en validation.
La synthèse artificielle de molécules a pris un rôle important dans notre société au cours du XXème siècle. Est-ce un bienfait? On peut en discuter sur le forum. Quoi qu'il en soit, la plupart de ces médicaments, parfums ou colorants sont des composés organiques, c'est-à-dire des molécules constituées principalement d'atomes de carbone reliés entre eux. Voici quelques exemples:
Morphine | $\alpha$-Ionone | Fluorescéine
La morphine1 est un anti-douleur naturel bien connu, l'$\alpha$-ionone2 est l'un des composants du parfum naturel de violette et la fluorescéine3 est une molécule artificielle qui émet de la lumière sous ultraviolets. Comme nous pouvons le constater, la structure de ces molécules repose bel et bien sur un squelette d'atomes de carbone. Le développement de nouvelles méthodes capables de former ces liaisons C-C est donc au cœur de la recherche en chimie organique. L'objectif de cet article est de donner un premier aperçu des couplages croisés catalysés par le palladium. Ces réactions apparues dans les années 1970 font désormais partie des "classiques" dans la formation de liaisons C-C et ont servi de base à tout un pan de la chimie moderne.
Les réactions catalytiques
Les couplages croisés catalysés au palladium sont des réactions dites catalytiques car elles fonctionnent grâce à un catalyseur. Un catalyseur est une espèce ajoutée dans le milieu réactionnel destiné à accélérer ou rendre possible une réaction chimique sans être consommé ni intégré au(x) produit(s). Parfois, le catalyseur peut même être récupéré à la fin de la réaction et réutilisé. Les réactions catalytiques sont classées en deux grandes familles: la catalyse hétérogène et la catalyse homogène.
La catalyse hétérogène
Dans ce type de catalyse, le catalyseur et le(s) réactif(s) ne sont pas dans la même phase (liquide, gaz, etc.). C'est par exemple le cas lorsque le catalyseur est un solide et que les réactifs sont des gaz, ou lorsqu'un catalyseur solide est mis en suspension dans une solution contenant les réactifs.
Un bon exemple de la vie de tous les jours est le pot catalytique4 qui équipe les voitures modernes. Il s'agit d'un filtre constitué d'un alliage métallique à travers lequel les gaz d'échappement passent. Au contact de l'alliage, plusieurs réactions chimiques sont accélérées, dont la réaction ci-dessous. L'objectif du pot catalytique est de convertir un maximum de CO en CO2 car le CO2 est moins nocif que le CO:
$2CO+O_2\rightarrow 2CO_2$
Pot catalytique
Un autre exemple est l'hydrogénation d'alcènes avec du palladium sur charbon Pd/C. Ce mélange de palladium et de graphite est une poudre noire plus ou moins fine que l'on met en suspension dans une solution contenant l'alcène à hydrogéner. Une pression d'hydrogène gazeux H2 est ensuite appliquée pour effectuer la réaction. Bien que cette méthode utilise du palladium, il ne s'agit pas d'un couplage croisé! Le palladium possède en effet de nombreux types de réactivité.
Hydrogénation avec Pd/C
La catalyse homogène
L'autre grande famille est la catalyse homogène, où le catalyseur et les réactifs sont dans la même phase. La plupart du temps, ils sont tous les deux dissous en solution.
Les couplages croisés catalysés par le palladium font partie de cette famille, comme nous le verrons dans la suite de cet article. Mais prenons déjà un autre exemple plus simple enseigné dès les premiers cours de chimie organique: l'estérification. Cette réaction bien connue est le mélange d'un acide carboxylique avec un alcool. La réaction peut fonctionner sans catalyseur, mais lorsqu'on ajoute un peu d'acide dans le milieu (H2SO4, HCl, etc.), elle est accélérée:
Estérification en milieu acide
Les couplages croisés
Passons au vif du sujet. Une réaction de couplage croisé permet de former une liaison entre deux fragments de molécules grâce à la présence d'un catalyseur métallique. Ces deux fragments ne réagiraient pas en l'absence du catalyseur. Si ces deux morceaux sont identiques on parle d'homocouplage, et s'ils sont différents de couplage croisé. À l'heure actuelle, il est possible de former toutes sortes de liaisons: C(sp2)-C(sp2), C(sp2)-N, C(sp)-(sp2), C(sp3)-C(sp3), etc. Cet article se focalisera au début sur les liaisons C(sp2)-C(sp2) puisque historiquement ce sont les premières à avoir été développées:
Liaisons C(sp2)-C(sp2)
Avant les années 1970, très peu de méthodes efficaces existaient pour former de telles liaisons directement. Puis sont arrivés les travaux de Kumada et Corriu5. Dès lors, l'intérêt pour les couplages croisés n'a cessé de grandir et des centaines d'articles scientifiques sont publiés chaque année sur le sujet, repoussant toujours les limites de ces réactions passionnantes.
Les ingrédients
Les trois ingrédients de base dans un couplage croisé traditionnel sont:
un réactif halogéné (R-X)
un réactif organométallique (R'-M)
un catalyseur au palladium (LnPd)
L'objectif d'un couplage croisé est de coupler un réactif halogéné avec un réactif organométallique. Voici quelques combinaisons possibles:
Ingrédients pour un couplage croisé
Les couplages croisés ont été développés pour une grande variété de réactifs organométalliques différents. À l'origine, Kumada a exploité les réactifs de Grignard (M = MgX). Ceux-ci sont simples à préparer mais ne tolèrent pas la présence de carbonyles à cause de leur réactivité bien connue avec ces derniers.6 Negishi7 a obtenu de meilleurs rendements pour les produits biaryles avec le zinc (M = ZnX2). Stille8 et Hiyama9 ont quant à eux développé de nouveaux réactifs basés sur l'étain (M = SnR3) et le silicium (M = SiR3). Finalement, l'un des meilleurs couplages est le fameux couplage de Suzuki10 qui utilise des acides boroniques ou des dérivés (M = B(OH)2, M = B(OR)2, etc.).
Le cycle catalytique
Les réactions citées ci-dessus suivent toutes la même séquence d'étapes élémentaires suivantes à quelques détails près: addition oxydante, transmétallation et élimination réductrice. Ces étapes constituent un cycle: la dernière étape régénère le système, lui permettant ainsi de répéter la séquence tant qu'il y a des réactifs à consommer. Voyons tout ceci en détail en étudiant le cycle catalytique général pour tout couplage croisé ainsi qu'un exemple particulier de couplage de Kumada catalysé par le complexe de palladium Pd(PPh3)4:
Cycle catalytique général | Exemple de couplage de Kumada
1) La première étape est l'addition oxydante (A): le composé halogéné s'additionne sur le palladium et fait passer son état d'oxydation de 0 à II. D'un point de vue mécanistique, on peut dire que Pd0 s'insère dans la liaison C-X du composé halogéné. Remarquons que dans notre exemple de couplage de Kumada, deux ligands PPh3 doivent préalablement se dissocier du complexe Pd0(PPh3)4 pour générer le catalyseur actif Pd0(PPh3)2.
2) La deuxième étape est la transmétallation (B): le groupe R' du composé organométallique R'M est transféré d'un métal à l'autre (de Mg à Pd dans le cycle de Kumada) et prend la place de l'halogène sur le palladium. L'halogène qui se trouvait sur le palladium est quant à lui éliminé avec le métal du réactif organométallique.
3) La dernière étape est l'élimination réductrice (C): les deux groupes R et R' sont éliminés pour former le produit de la réaction tout en régénérant le catalyseur. L'élimination est dite réductrice car le palladium passe de II à 0.
Conformément à la définition d'une réaction catalytique, le catalyseur est régénéré à la fin de la réaction. Ainsi, pour 1 mole de réactifs, il n'est pas nécessaire de charger 1 mole de catalyseur: 0.1 mole ou moins peuvent suffire. Certaines réactions industrielles tournent même avec 0.00001 mole de catalyseur par mole de réactifs, c'est-à-dire que chaque molécule de catalyseur fait 100'000 cycles au cours de la réaction!
Ces trois étapes constituent la base de toutes les réactions de couplages croisés mais de petites variations peuvent exister. La réaction de Suzuki requiert par exemple une base tandis que la réaction de Hiyama nécessite une source de fluorure. Je vous encourage à jeter un coup d'œil à ces autres cycles catalytiques dans un livre ou sur internet, vous verrez que le principe est toujours le même.
La course à la performance
Au cours de ces 40 dernières années, beaucoup de recherches ont été menées pour mieux comprendre chaque étape et ainsi optimiser la réaction. Premièrement, l'efficacité de l'addition oxydante dépend de l'halogène: l'insertion du palladium 0 dans une liaison C-I est beaucoup plus facile que dans une liaison C-Cl, tout simplement car l'énergie de liaison est plus faible. Quant aux liaisons C-F, elles ne peuvent normalement pas subir d'addition oxydante.
Liaison
Énergie de liaison
C-I
238 kJ/mol
C-Br
285 kJ/mol
C-Cl
330 kJ/mol
C-F
485 kJ/mol
Comme alternative aux halogènes, il est possible d'utiliser les triflates C-OTf. Ceux-ci sont assez facile à préparer et sont très réactifs (ils se situent entre les C-I et les C-Br). Plusieurs réactions permettent de les faire, en voici juste un exemple:
Préparation d'un triflate
Le choix de l'halogène est important, mais l'axe d'optimisation le plus important est sans aucun doute la modification des ligands du catalyseur. Au-delà de la simple triphénylphosphine PPh3 que nous avons vue, des centaines d'autres phosphines sont désormais dans le commerce: liste de phosphines. Impressionnant n'est-ce pas?
Plusieurs tendances ont été constatées. Premièrement plus les phosphines sont riches en électrons plus l'addition oxydante se fait facilement (ex: PtBu3). C'est logique: le palladium est mieux armé (plus nucléophile) pour s'insérer dans la liaison C-X. Pour accélérer l'élimination réductrice, les "gros" ligands (encombrés stériquement) sont meilleurs. L'encombrement favorise l'élimination du produit pour que les ligands se sentent à nouveau plus libres autour du palladium. Il existe ensuite les ligands bidentés (plus stables, ex: dppf), les ligands chiraux (pour les réactions énantiosélectives, ex: (S)-iPr-PHOX), etc. etc. Parmi les ligands couramment utilisés on rencontre le XPhos car il cumule plusieurs de propriétés mentionnées ci-dessus.
Ligands courants
Comment s'y retrouver parmi tous ces ligands? En général, lorsqu'on veut faire un "simple" couplage, on commence par tenter la réaction avec un catalyseur classique tel que le Pd(PPh3)4 ou le Pd(dppf)Cl2. Si ça ne fonctionne pas, on cherche alors quelque chose de plus sophistiqué en fonction de ce qu'on a obtenu comme produits/sous-produits.
De la théorie à la pratique
Avant de continuer, passons à un chapitre un peu plus récréatif: des applications réelles de la réaction de Suzuki. La première provient de mon cursus tandis que la seconde a été publiée par une entreprise pharmaceutique dans le cadre du développement d'un médicament potentiel contre certaines maladies pulmonaires.
Expérience personnelle
Voici un couplage de Suzuki effectué pour préparer le nitrobenzène substitué 3, un composé dont j'avais besoin. Il existe certainement d'autres conditions réactionnelles ou méthodes pour synthétiser cette molécule, mais la réaction employée dans le cas présent s'est avérée efficace et a fourni le produit avec un bon rendement de 91%:
Couplage de Suzuki
Quels sont les points particuliers à prendre en compte pour concevoir le montage? Il y en a principalement deux:
La température à atteindre est de 100°C. Le point d'ébullition du dioxane étant de 101°C, il faut utiliser un réfrigérant et effectuer la réaction à reflux pour éviter que le solvant ne s'évapore.
La réaction est sensible à l'oxygène O2 car celui-ci peut potentiellement oxyder le catalyseur actif Pd0 en PdII et le désactiver. Il faut ainsi effectuer la réaction sous atmosphère inerte.
Montage sous atmosphère inerte | catalyseur Pd(dppf)Cl2
Une fois la verrerie assemblée, il ne reste plus qu'à lancer la réaction. Le catalyseur, l'ester boronique, le dioxane, la solution aqueuse de NaOH 3 M puis le triflate sont chargés dans le ballon. Le tout est mis sous atmosphère inerte d'azote et chauffé à 100°C pendant une nuit. Après extraction, concentration et purification par chromatographie sur colonne, le produit final est obtenu avec 91% de rendement.
Comme vous pouvez le constater, il n'y a rien d'insurmontable. Ce couplage a bien fonctionné dès le premier essai, mais ce n'est pas toujours le cas: il faut parfois varier plusieurs paramètres avant de rencontrer le succès. Tout est question d'expérience (et de chance!).
Application industrielle
Ces réactions ne sont pas uniquement utiles en milieu académique sur quelques dizaines de milligrammes, elles sont également largement exploitées dans l'industrie de la chimie fine. En 2011, un article qui recense des dizaines d'exemples de couplages croisés utilisés à grande échelle a été publié dans la revue Chemical Reviews.12 Comme promis, en voici un qui a servi pour obtenir un candidat potentiel contre certaines maladies pulmonaires:13
Couplage de Suzuki
La réaction a fourni 60 g de produit avec un très bon rendement de 88%. Il est intéressant de noter que le couplage s'est produit entre B(OH)2 et Cl (couplage croisé) et non entre B(OH)2 et F (homocouplage non souhaité), ce qui démontre en partie que l'addition oxydante se fait préférentiellement avec les halogénures dont l'énergie de liaison C-X est la plus faible!
Quelques réactions dérivées
En parallèle aux réactions que nous avons vues jusqu'ici, qui permettaient essentiellement de former des liaisons C(sp2)-C(sp2), d'autres réactions similaires ont été développées.
Historiquement la réaction de Heck a été l'une des premières réactions de couplage croisé. Elle forme également une liaison C(sp2)-C(sp2), mais en se passant du réactif organométallique. Elle permet le couplage d'un réactif halogéné avec une double liaison. Elle est donc extrêmement utile en synthèse totale puisqu'on peut alors facilement attacher des fragments sur toute molécule possédant une double liaison. Le cycle catalytique est quelque peu différent, je vous laisse le consulter vous-même:14
Couplage de Heck
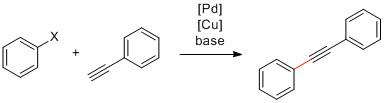
Passons à un autre type de liaison: C(sp2)-C(sp). Il s'agit du couplage de Sonogashira qui permet d'attacher un alcyne terminal sur un carbone hybridé sp2. Dans cette réaction, un co-catalyseur de cuivre est utilisé pour former du réactif organométallique cuivré in situ à partir de l'alcyne terminal au fur et à mesure de l'avancement de la réaction:15
Couplage de Sonogashira
Finalement, une réaction synthétiquement intéressante est celle de Hartwig-Buchwald puisqu'elle forme une liaison C-N.16 En général les ligands bidentés ou encombrés stériquement sont à privilégier pour éviter la formation de sous-produits non-désirés. Cette méthode surclasse la simple substitution aromatique nucléophile sans catalyseur. Cette dernière ne fonctionne en effet qu'avec des halogénures d'aryles très pauvres en électrons et est très limitée.
Couplage de Hartwig-Buchwald
Les activations C-H
Les activations C-H sont des réactions très récentes et prometteuses. Un réactif possédant une simple liaison C-H peut y être utilisé à la place du réactif halogéné! On utilise le terme activation car la liaison C-H en question est considérée comme intrinsèquement non-réactive et doit être activée. C'est par exemple le cas des liaisons C-H du benzène ou du pentane. En revanche, une liaison C-H en position $\alpha$ d'une cétone est considérée comme réactive puisqu'on peut facilement la casser par déprotonation et n'entre donc pas en considération dans ce chapitre.
Pour comprendre ce que signifie activation, rappelons ce qu'est une réaction bimoléculaire en solution: ce sont deux réactifs qui nagent dans un solvant et entrent en collision aléatoirement. Toutes ces collisions n'aboutissent pas forcément à une réaction, il faut que la collision soit efficace: avec suffisamment d'énergie et selon la bonne orientation des molécules. Or, entre un catalyseur au palladium et une liaison C-H d'un benzène, il faudra peut-être des milliards de collisions avant que le palladium ne s'insère dans la liaison car celle-ci est trop résistante. En d'autres termes, la réaction ne fonctionnera pas.
Dans une réaction d'activation C-H, puisque les collisions entre le catalyseur et la liaison C-H ne sont pas assez souvent efficaces, la stratégie consiste à diriger le palladium proche de la liaison C-H et le forcer à réagir avec. Pour ce faire, il faut que le réactif portant la liaison C-H porte aussi un groupe directeur à proximité pour "ancrer" le palladium tout près de la liaison à attaquer. Ainsi, il tentera sans relâche de la rompre, jusqu'à réussir.
Il existe une multitude de réactions de ce type: arylations, alkylations, oléfinations, etc. Leurs mécanismes pouvant fortement différer, ne considérez donc pas l'exemple choisi ici comme une généralité! Intéressons-nous à l'arylation C-H en position ortho. Plusieurs groupes fonctionnels peuvent servir de groupes directeurs. Parmi les plus simples on trouve les amides, les acides carboxyliques ou les 2-pyridyles:
Activations C-H avec groupes directeurs
Voici le cycle catalytique proposé pour cette réaction:17
Cycle catalytique de l'arylation C-H en ortho
L'une des différences avec le cycle étudié auparavant est l'état d'oxydation du palladium, qui varie entre II et IV au lieu de 0 et II. Il y aurait énormément de choses à expliquer concernant les états d'oxydation du palladium dans les couplages croisés, mais laissons de côté ceci car ce n'est pas l'objectif de l'article et concentrons-nous plutôt sur le rôle du groupe directeur. Grâce à celui-ci, le catalyseur PdII(OAc)2 peut activer et rompre l'une des liaisons C-H en position ortho du groupe directeur. Ensuite, une addition oxydante suivie d'une élimination réductrice délivre le produit. Finalement, pour régénérer le catalyseur, de l'acétate d'argent est utilisé: les ions Ag+ ayant une forte affinité avec les halogénures, ils arrachent facilement ces derniers du palladium.
Ces activations C-H sont donc très attractives lorsque le produit que l'on souhaite synthétiser porte déjà un groupe utilisable comme groupe directeur car celui-ci permet l'économie d'une liaison C-X. Et ceci n'est que le début de l'ère des activations C-H! Pour vous en convaincre, vous pouvez jeter un coup d'œil aux publications du groupe de recherche de Jin-Quan Yu: activations CH en position meta, activations de liaisons C(sp3)-H, réactions énantiosélectives, etc.!
Le Prix Nobel de chimie 2010
Le développement de toute cette chimie méritait bien un Prix Nobel. Ce fût chose faite en 2010 lorsque la récompense fût attribuée conjointement à Richard F. Heck, Ei-ichi Negishi et Akira Suzuki, soit trois grands chimistes parmi les pionniers du domaine:18
"pour les couplages croisés catalysés par le palladium en synthèse organique"
Conclusion
Apparus dans les années 1970, les couplages croisés catalysés par le palladium sont devenus des réactions centrales en chimie organique et sont désormais massivement utilisés tant en milieu académique qu'en industrie. À l'heure actuelle, des milliers de groupes de recherche à travers le monde travaillent sur ce sujet.
Vous l'aurez compris, cet article n'est qu'une modeste introduction à tout ce pan de la chimie et avait pour objectif de présenter les concepts de base puis de donner envie d'en apprendre plus. De nombreux thèmes n'ont donc pas pu être abordés. La principale omission est certainement que le palladium n'est plus l'unique métal de transition exploitable: des couplages croisés avec du nickel, du rhodium, du ruthénium ou même du fer ou du cobalt ont été développés entre-temps. Ces deux derniers sont particulièrement intéressants en terme de coût puisque ce sont des métaux bon marché.
Bref, il y aurait encore beaucoup à dire, mais arrêtons-nous là! N'hésitez pas à commenter et à venir poser toutes vos questions sur le forum Sciences.
Je suis un mega noob en chimie. Pire, je déteste ça (du moins, celle que j'ai faite à l'école, c'est-à-dire celle que j'ai faite tout court).
Est-ce un bienfait?
Une habitude suisse ? Chez nous, on fait précéder le point d'interrogation d'une espace.
Voici quelques exemples:
De même.
La morphine1 est un anti-douleur naturel bien connu, l'α-ionone2 est l'un des composants du parfum naturel de violette et la fluorescéine3
Il serait préférable de mettre les liens directement sur les mots concernés.
émet de la lumière sous ultraviolets
Ca signifie qu'elle en émet quand elle est placée sous des rayons UV ou qu'elle émet des UV ? Je dirais la première, mais j'ai un petit doute quand même.
partie des "classiques"
Je mettrais des guillemets français.
Les réactions catalytiques
une espèce ajoutée dans le milieu réactionnel destiné
e
deux grandes familles:
Espace.
La catalyse hétérogène
le catalyseur est un solide et que les réactifs sont des gaz
Une note fournissant un exemple serait appréciée.
catalyseur solide est mis en suspension dans une solution contenant les réactifs
De même.
Quand tu dis "en suspension", c'est vraiment en suspension ou c'est un terme technique pour dire "solide dans liquide" ?
moins nocif que le CO:
Espace.
il ne s'agit pas d'un couplage croisé!
Idem.
Le palladium possède en effet de nombreux types de réactivité.
Je ne comprends pas cette phrase. Mais je n'ai pas de connaissances poussées en chimie non plus.
La catalyse homogène
cours de chimie organique:
Espace.
elle est accélérée:
Idem.
Les couplages croisés
Une réaction de couplage croisé permet de former une liaison entre deux fragments de molécules grâce à la présence d'un catalyseur métallique.
Un exemple simple d'utilisation serait le bienvenu.
sortes de liaisons:
Idem.
à avoir été développées:
Plutôt un point.
Les ingrédients
traditionnel sont:
Espace.
Voici quelques combinaisons possibles:
Idem.
Le cycle catalytique
à quelques détails près:
Espace.
addition oxydante, transmétallation et élimination réductrice
Une liste serait plus claire.
constituent un cycle:
Espace.
la dernière étape régénère
Répétition : la dernière régénère
palladium Pd(PPh3)4:
Espace.
1) La première étape
Si tu mets un point après le chiffre, ça crée une liste. A voir si ça ne rendrait pas mieux.
l'addition oxydante (A):
est la transmétallation (B):
est l'élimination réductrice (C):
Espaces.
Ainsi, pour 1 mole de réactifs, il n'est pas nécessaire de charger 1 mole
une mole
de catalyseur:
Espace.
c'est-à-dire que chaque molécule de catalyseur fait 100'000 cycles
Pas trop compris cela.
au cours de la réaction!
Espace.
ou sur internet, vous
Internet : vous
La course à la performance
Je m'arrête là pour aujourd'hui. Probablement tout court en fait : je crains de ne pas avoir le niveau requis pour comprendre la suite. Je verrai ce qu'il est.





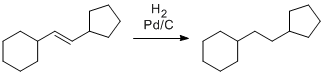

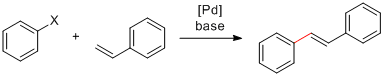



 ), bref, c'est bien quoi
), bref, c'est bien quoi