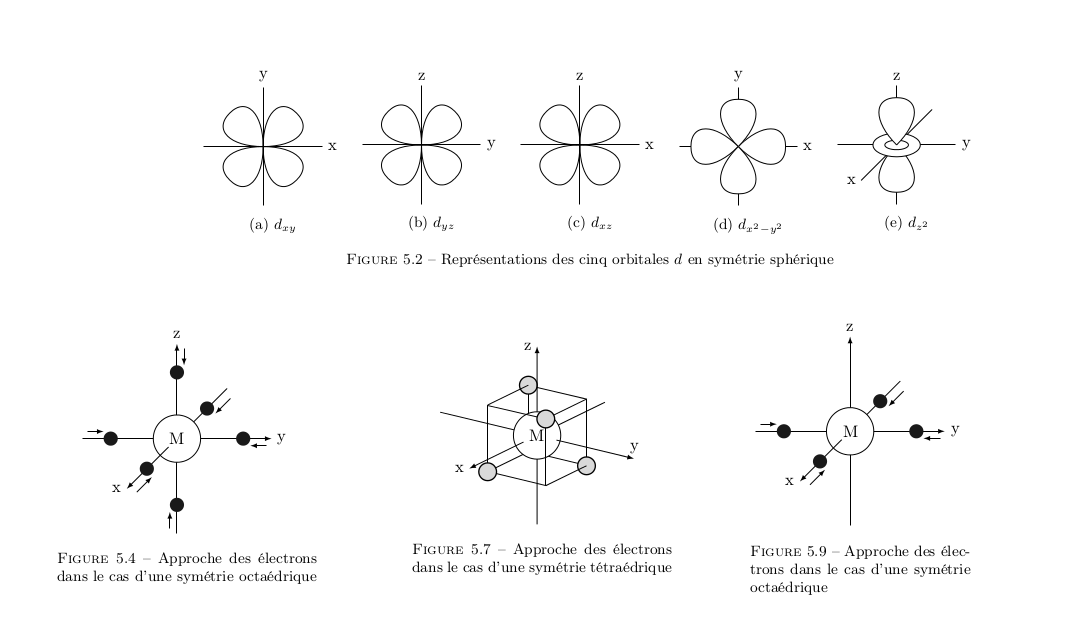
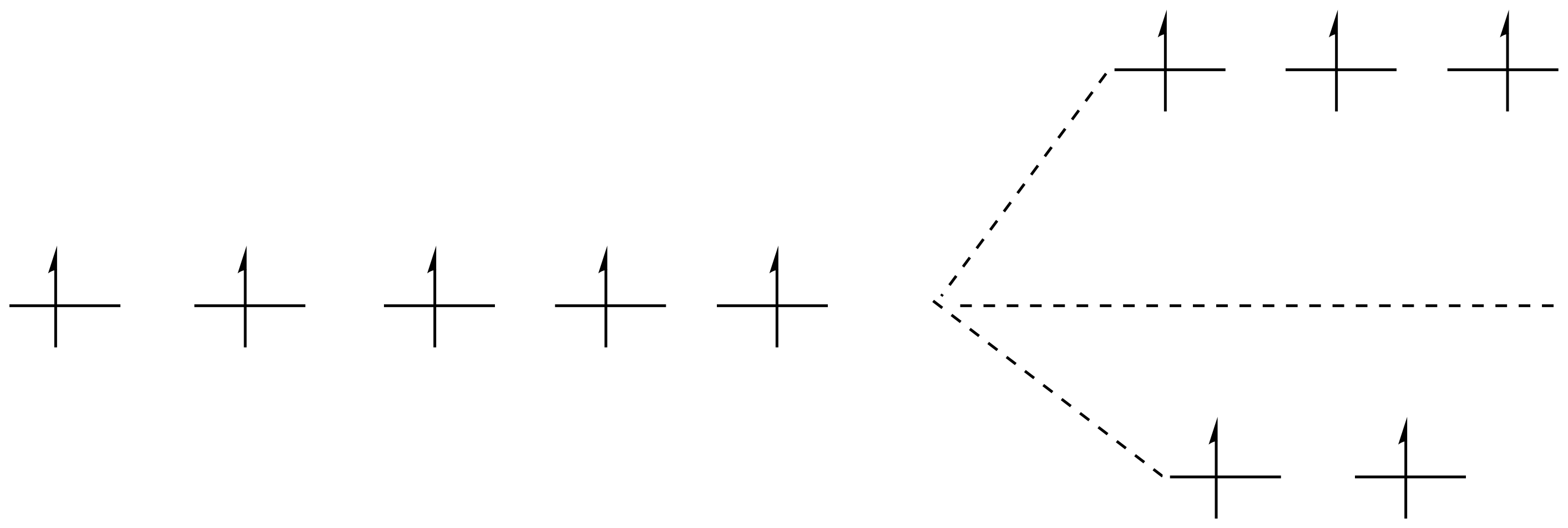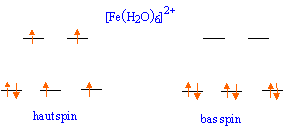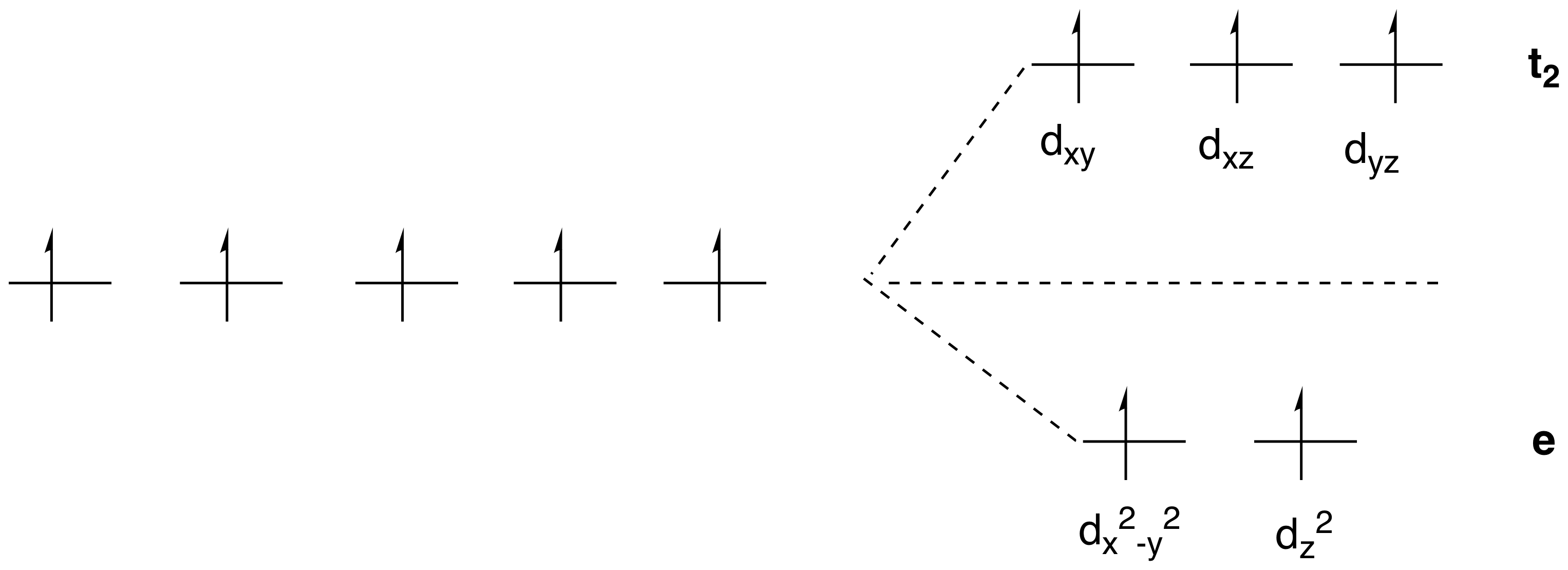On m’avait dis de regarder la théorie du champ cristallin quand j’avais ouvert un topic sur les complexes.
J’ai donc un peu regardé cette théorie mais j’ai pas super bien compris.
Ce que j’ai compris c’est que les métaux de transitions ont leurs électrons dans leurs orbitales d avec différents niveaux d’énergie (pourquoi ?).
Aussi, la différence d’énergie entre ces orbitales d dépend de la nature du ligand. Je crois qu’il augmente plus le ligand est bon (?). Est-ce qu’il dépend aussi du métal par hasard (Mn est-il différent de Fe?) ?
Si vous pouviez déjà m’expliquer un peu plus en détails pour le complexes tétraédriques Td (on dirait que ce sont les plus simples )
Puis on m’a aussi parlé de haut spin et bas spin mais j’ai pas trop d’idées de quoi il s’agit.
Bas Spin = Champ fort (série spectrochimique, ligands fort = $CO$, $CN$)
Haut Spin = Champ faible (ligands faible = $H_2O$, $Cl$)
En gros un ligand de bas spin, à champ fort donc va forcer les électrons célibataire du métal IMC à se couplet en DNL. Donc si tu as un métal ayant 5 électrons célibataires, tu peux te retrouver à tous les apparier pour en faire un complexe d’un certaine hybridation (d’une certaine géomètre…)
Les Orbites :
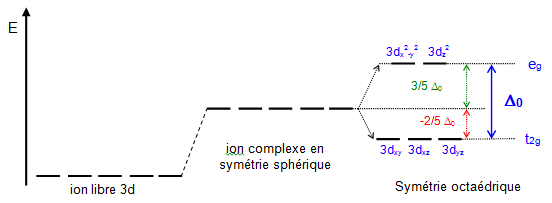
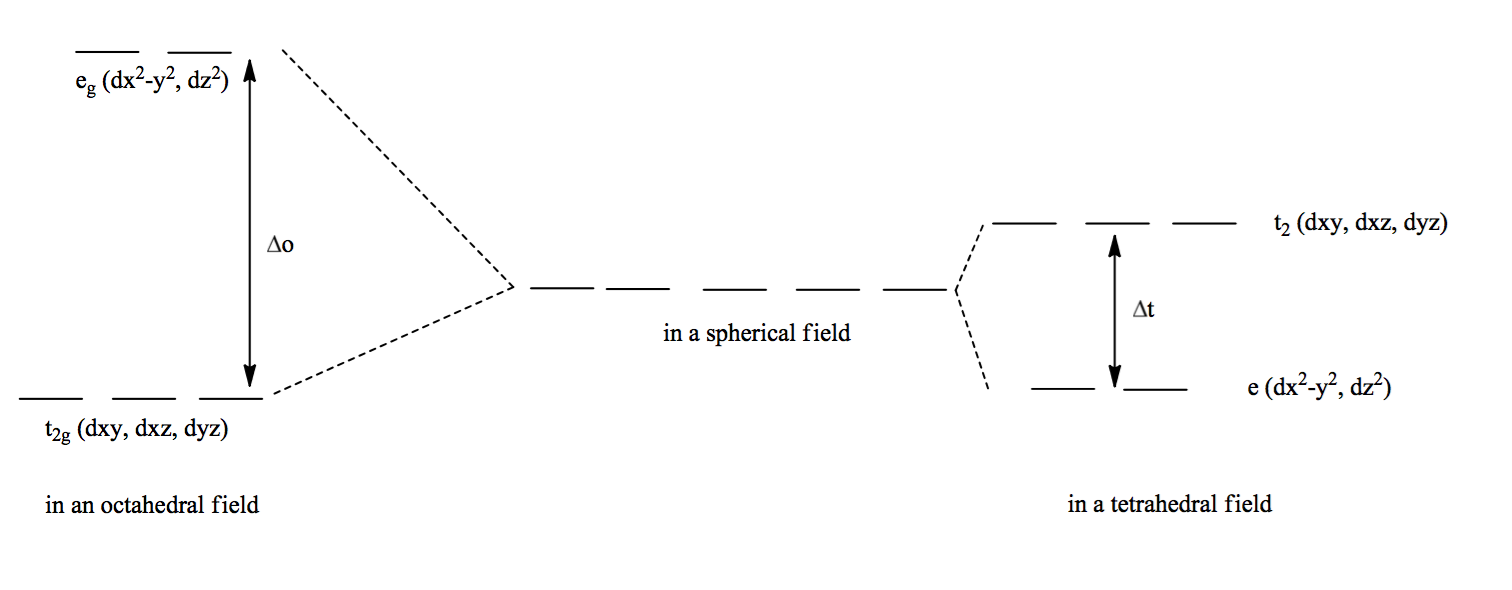
La théorie du champ cristallin s’explique avec les orbitales $t_{2g}$ et les orbitales $e_g$ quand tu es en octaédrique$\Delta_O$. Et en $t_2$ et $e$ pour tétraédrique$\Delta_{t}$.
$$
\frac49\Delta_O = \Delta_t
$$
Ce $\Delta$ est directement lié à la différence d’énergie entre les sous-orbitales générés par les ligands. En somme ton métal possède 5 cases quantiques $3d$ à priori. Et c’est cette orbitales là qui va se faire influencée. Déjà lorsqu’un métal se retrouve en solution, il y a une sphère de coordination qui la destabilise :
Diagramme d’Orbitale d’un métal de transition typique (formation de couches octaèdriques)
Une fois déstabilisée, tu vois que les orbitales se scindes en 2 parties. C’est là que tu vas avoir une répartition différentes selon la géométrie du complexe que tu as. Comme l’indique la légende que j’ai placée sous l’image ici tu est en Octaédrique.
Geometries
Nombre de $e$
type d’orbitale haute
Nombre de $t$
type d’orbitale basse
Octaédrique
$2$
$e_g$
$3$
$t_{2g}$
Plan Carrée
$2$
$e_g$
$3$
$t_{2g}$
Tetraèdrique
$3$
$t_{2}$
$2$
$e$
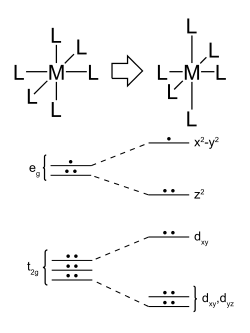
Avec la subtilité que le Plan Carré est en fait une déstabilisation (again again, and again ) des sous-couches octaédriques.
Destabilisation des sous-couches $t_{2g}$ et $e_g$
Car si tu enlève les orbitales responsables de la liaison dans l’axe de la hauteur, tu ne garde que les 4 équatoriales. Ce qui revient en terme d’orbitales à la placer de manière inaccessible (hors de portée énergétiquement) : très hautes. Cela s’appelle l’effet Jahn-Teller. Tu vois que le complexe devient "étiré" lorsque l’on déplace les sous-couches. L’orbitale responsable de ce lien transverse entre une partie haute et une partie basse dans un complexe est l’orbitale $z^2$ je crois, car elle se désactive de pars et d’autre.
Orbitale $d_{z²}$ responsable de l’effet Jahn-Teller
Elle peut-être rendu inaccessible, alors que les orbitales du genre $d_{xy}$ sont responsable du plan carré :
Orbitales $d_{xy}$ responsable du plan carré
Après là on va vraiment à la frontière de ce que je sais sur les complexes et leurs théorie en terme de Chimie Quantique. Faudra demander à Pierre_24 de faire des $H$ opérateur Hamiltoniens appliqué à la spectroscopie pour nous
L’Energie d’éclatement du réseau cristallin $\Delta_O$, ses influences et la spectroscopie
Maintenant la partie la plus rigolote $\Delta_O$ est directement relié à la couleur d’un complexe. Alors il faut une petite formule pour permettre à $\lambda$ d’apparaître :
$$
\Delta_O = \frac{N_Ahc}{\lambda}
$$
De cette formule tu peux déduire, à partir d’une couleur monochromatique émise par un complexe, quel est l’énergie de ce dernier. (d’après moi s’grave stylé)
Exemple de complexe que j’ai synthétisé.
Attention toutefois à prendre la couleur absorbé pour la formule suscitée. Car la couleur emise est son inverse. "Notion de couleur complémentaire"
Spectre des couleurs, avec en face de chacune leur couleur complémentaire
On a aussi pour habitude d’utiliser la valeur $\sigma_O$ pour definir l’energie du complexe. Mais attention les formules ne sont pas les mêmes, vue que $\sigma_O$ s’exprime en $cm^{-1}$ faut pas tout mélanger lorsque tu compare.
$$
\sigma_O = \frac1\lambda
$$
Donc on vient de voir que le $\Delta_O$ symbolise beaucoup de chose dans les complexes. Et même plus que tu ne le penses. En fait ce dernier est totalement relatif aux métaux et aux ligands qui vont être employés. C’est donc un indice absolue pour définir la stabilité d’un complexe. Je ne vais pas te faire un TD la dessus, mais on pourrait s’amuser a donner des résolutions d’exercice un jour ?
Les facteurs qui influencent $\Delta_O$ sont nombreux :
Le Ligand
L’IMC
Le Solvant
Le Ligand est vraiment le plus dominant. Il va vraiment pouvoir te permettre de juger de la stabilité d’un complexe à peu de chose près. Peu importe l’IMC. cf : série spectrochimique
Pour mieux comprendre les facteurs qui vont modifier la stabilité d’un complexe par rapport à un métal c’est compliqué… Pour si peu de résultat c’est pas simple. Certains essayent de vulgariser la mnière dont la stabilité evolue mais… la réalité c’est qu’il faut voir du coté de la $HSABT$ (Hard and Soft Acide-Base Theory). En gros cette théorie explique pourquoi $NaOH$ et $KOH$ n’ont pas le même $pK_a$ alors que l’anion est le même $HO^-$. C’est dur de vouloir le diagnostiquer "avec les mains". (Si le noyau est gros et est beaucoup chargé, genre $Co^{3+}$ ça peut être suivi d’un grand $\Delta_O$)
Merci beaucoup pour ces premières infos. Je vais déjà te répondre avec mes questions.
Si on prend le même exemple que dans mon post d’avant, MnCl4. C’est un complexe tétraédrique (plan carré?). Si je dessines les électrons d du métal (Mn2+ donc) on aurait quelque chose du genre:
c’est bien ça ? Avec le niveau du haut appelé $t_2$ et celui du bas $e$. Je suppose que $\Delta _t$ c’est la différence d’énergie des deux niveaux ?
Comme il est tétraédrique, il est haut spin (mais encore ça veut dire quoi vraiment?). J’ai vu sur Wikipedia que tous les complexes tétraédriques sont haut spin. Possible ?
Est-ce que chaque "case" a un nom spécifique genre en fonction de l’orientation de l’orbitale ou je ne sais quoi?
Ca paraît bizarre car ça voudrait dire CuCl4, CoCl4 ou MnCl4 seraient équivalents et tous haut spin peut importe le métal au milieu ?
Edit: J’ai pas encore lu ton message, on écrivait en même temps. Y’a peut-être mes réponses
Oui j’redigais mon message en ce moment même. Hier j’ai donné 2-3 pistes… Mais je rentré de soirée j’étais pas des plus "clair" lol.
Alors attention un complexe plan carré et tétraédrique ne sont pas les mêmes du coups ! Je te parlerai bien volontiers des hybridations dans ce post, faudra juste être patient que je re-edite ce dernier.
Mais en gros, comme en chimie organique, un tétraèdre résulte d’une hybridation $sp^3$ héhé. Et les orbitales mises en jeux ne sont pas positionné pareil, tu vois bien que le plan carré c’est un effet Jahn-Teller qui doit apparaître sur ton dessin.
L’effet du Ligand sur les electrons
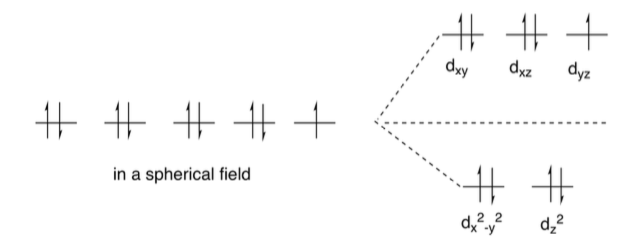
Les électrons se placent, sur un diagramme d’orbital moléculaire, d’abord sur les orbitales de plus basse énergie, et selon cet règle (Règle de Hund) tu ne devrais pas avoir, sur ton schéma, 5 electrons celibataires.
Normalement sur des orbites de plus petite énergie tu as un couplage des électrons ensemble.
Exemple d’appariement des electrons
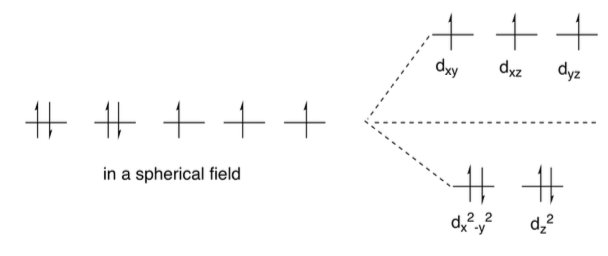
Et ces agencements, savoir si tu respecte la règle de Hund ou non, c’est la force du ligand qui va te le dire. Tu vois maintenant à quoi correspond visuellement un Ligand à champ fort (bas spin). Dans ce cas précis que tu vois sur l’image tu sais que l’eau n’est pas un très bon ligand donc il y a fort à parier que c’est le diagramme de gauche le plus vraisemblable. Celui où les electrons occupe un max d’espace.
Hybridation
Imaginons ce coups-ci que nous ayons $[Fe(CN)_6]^{3-}$, un IMC chargé $+3$ et des ligands qui sont très fort ? Ca promet de donner la configuration en bas spin, diagramme de droite ici.
$$[Fe^{+3}] : [Ar]3d^6$$
On sait que nos ligands, quand ils débarquent pour faire une liaison dative (de coordination) il donne un doublet pour combler une case quantique vide.
On sait aussi que dans la configuration bas spin il reste 2 cases vides à combler.
Donc on peut écrire :
$3d$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
Avec les deux dernières pairs d’electrons relatives à ligands $CN^-$ notons les $d^2$
Donc il reste $4$ ligands à placer, mes nos orbitales $3d$ sont pleines que faire ?
On va se déporter sur la suite de Klechkowski :
$$[Fe^{+3}] : [Ar]3d^6 4s^0 4p^0$$
$3d$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
$4s$
$\uparrow \downarrow$
$4p$
$\uparrow \downarrow$
$\uparrow \downarrow$
$\uparrow \downarrow$
Avec les $d^2 + s^1 + p^3$ remplis par les $6$ ligands $CN^-$
On lit l’hybridation alors : $d^2sp^3$ et ça c’est relatif, direct cash yolo upercut, à la géométrie de la molécule ! $d^2sp^3$ ou $sp^3d^2$ ça signifie octaédrique.
Pour expliquer pourquoi certaines orbitales étaient hautes en énergie et d’autre basses, mon cours avait une méthode assez explicite, à défaut d’être 100% réaliste.
Champ cristalin
On réduit le liguant à un seul électron, on considère uniquement les orbitales $d$ du métal, et on fait comme si les électrons se repoussaient. Par exemple, dans le cas de la symétrie octarédrique, les orbitales plus hautes en énergie seront celles qui prendront les électrons directements dans leur lobes, donc $d_{x^2-y^2}$ et $d_{z^2}$, tandis que les trois autres orbitales seront abaissées en énergie, car l’électron ira se loger "entre les lobes".
À l’inverse, en symétrie tétraédrique (pas carré plan), c’est les orbitales "bisectrices" ($d_{xy}$, $d_{yz}$ et $d_{xz}$) qui seront plus énergétiques car les électrons viendront directement "vers les lobes", alors que les deux autres orbitales seront abaissées en énergie. Finalement, pour la symétrie tétragonale, c’est le bordel, et je (me) cite:
Il y a deux manière de raisonner. La première est de considérer, comme cela a été fait avant, l’approche de quatre ligands sur les axes $\pm$x et $\pm$y, mais en raisonnant orbitale par orbitale :
Tout d’abord, la pire des 5, $d_{x^2-y^2}$, qui possède des orbitales justement sur ces axes, d’où le niveau énergétique le plus élevé, qu’on nommera $\ce{b_{1g}}$.
nsuite, il y a l’orbitale $d_{xy}$, avec lequel les interactions seront plus faible : il s’agira donc d’un niveau d’énergie intermédiaire, mais déstabilisant, qu’on notera $\ce{b_{2g}}$.
Enfin, l’interaction avec l’orbitale $d_{z^2}$, et en particulier sa zone centrale, son beignet sera jugée faible, et notée $\ce{a_{1g}}$.
Il reste $d_{yz}$ et $d_{yz}$, avec lesquelles l’interaction est la plus faible, et reprises dans le groupe $\ce{e_g}$.
(Quant à l’autre manière de raisonner, elle est de partir de la symétrie octaédrique et d’enlever les ligands situées en $\pm$z)
Comme je dit, c’est pas fondamentalement correct (densité de probabilité de présence, toussa), mais ça fait le taf. Pour aller plus loin, malheureusement, il faut inclure les orbitales moléculaires, c’est sympa mais c’est long à expliquer comme ça (par contre, ça explique la série spectrochimique).
Merci beaucoup pour toutes ces explications qui sont très détaillées. Il y a toujours quelque chose que je comprends pas bien, c’est l’influence du métal (d’ailleurs IMC c’est pour quoi?).
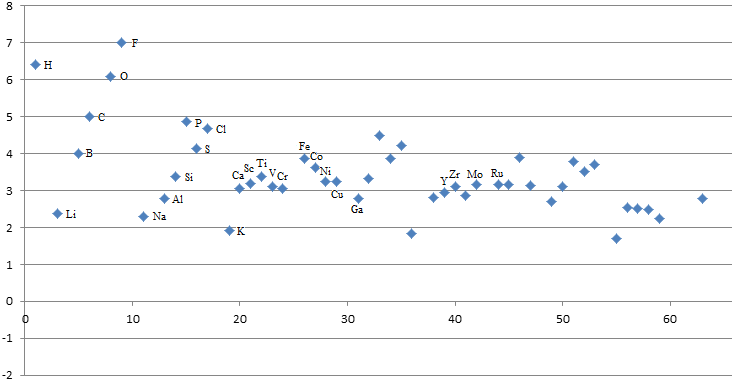
De ce que je comprends, la différence d’énergie est plus grande pour un métal 3+ que 2+ (par exemple Co3+ > Mn2+). Par contre, j’avais lu qu’on avait aussi quelque chose comme ça: Mn2+ < Ni2+ < Co2+ . Je sais pas si c’était une source fiable mais comment on peut expliquer ça ? D’ailleurs, le cuivre II il se situerais où?
Depuis plusieurs messages où tu parles des Complexes de Transitions j’utilise des fonctionnalités de ZDS, la balise mathématiques et les balises de citations, et même les balises pour les abréviatifs.
Quand tu constate un acronyme inconnu, et qu’il est souligné en pointillé cela signifie qu’une définition plus exacte est donné par le rédacteur dans son message. Au survole du curseur sur le mot la définition s’affiche.
La liste que tu essaye de mettre au point se réfère à $HSABT$ donc tu peux relire mon précédent paragraphe pour être sur d’avoir saisi. Sinon cadeau et gift
D’accord mais je suis moyen convaincu car si on lit la page Wikipedia en anglais que tu m’as donné, on voit que Mn, Fe, Co, … sont des métaux mi-moux / mi-durs et il y a pas réellement de classification entre ceux-ci donc dire Mn2+ < Ni2+ <… ne fait pas de sens.
Bah comme dis il y a des formules, ça se calcul en fait. Lire une phrase qui te dis que "des fois c’est pas simple" ça veut pas dire que certains modèles ne te donne pas de bonnes réponses.
Ordonné (Dureté en eV) Abscisse (Numéro Atomique u.a.)
Edit : Sinon le top pour savoir si un complexe est stable c’est de calculer $\dfrac1\beta = K_d = \Pi \left( a_{i}^{\nu_i}\right)$
OK ! Dernière question: un composé est toujours haut spin ou bas spin ou bien il peut être ni l’un ni l’autre ?
S’il y a qu’une configuration possible par exemple dans Ni2+ c’est ni l’un ni l’autre ou je me trompe? De plus, est-ce qu’on dis que l’IMC est haut / bas spin ou le complexe ?
Si on en revient à [MnCl4]2- est-ce que tu es d’accord que le diagramme ressemblerait à ça ? Je suis parti du principe que c’était tétraédrique mais j’ai toujours pas compris comment faire la différence entre tétraédrique et plan carré (plutot prévoir à l’avance… NiCl4, MnCl4, CuCl4, CoCl4, MCl4 où M = métal plus généralement pour moi c’est tétraédrique mais c’est sûrement faux…).
Juste pour dire que y’a pas de règle pour dire si un complexe est tétraédrique ou plan carré (ça se calcule, mais ça fait appel à la chimie quantique, donc disons qu’il n’y a pas de règle simple).
Juste pour revenir un peu au dessus, la théorie dur/mou fonctionne relativement bien dans ce genre de cas, par contre ça n’explique pas le pourquoi : sans entrer dans les détails, parce que c’est une question complexe (ahah ) ça a un rapport avec le fait d’avoir des orbitales capables de faire des liaisons pi. Ce qui, du coup, reviens effectivement à une histoire de Gap homo-lumo
Juste pour dire qu’effectivement c’est pas facile à comprendre le pourquoi sans rentrer dans des calculs de fous (et approchés…).
@Blackline > T’es sûr que pour un complexe tétraédrique il y a 3 $e$ et 2 $t_2$ ? Il me semblait que c’était le contraire. En tout cas, voici une capture d’écran de ce que j’ai dans un de mes cours.
Crystal Field Theory for Tetrahedral Complexes
Et oui, l’IMC peut être ni haut spin ni bas spin s’il n’y a qu’une configuration possible.
Du coup, s’il n’est ni l’un ni l’autre, on en déduire que le complexe n’est pas tétraédrique (vu qu’on a la propriété que tous les tétraédriques sont haut spin) ?
Je reviens à la charge car je me suis amusé à dessiner suivant le schéma de ZDS_M plusieurs diagrammes d’orbitales d de certains métaux en les considérant tétraédriques.
Par exemple:
Diagramme orbitales d - Cu2+
Ici par exemple je sais pas dire si c’est haut ou bas spin. J’aurais dis haut car c’est tétraédrique mais d’un autre côté c’est la seule configuration possible (ou mettre un électron dans d-yz mais c’est équivalent vu que c’est la même énergie) donc ça serait haut ou bas (dans le sens ça revient au même) ?
Pareil pour Co2+ j’ai ça :
Diagramme pour Co2+
et encore une fois y a pas deux possibilités (vu qu’on met un électron dans chaque case d’abord quand c’est au même niveau d’énergie).
Du coup, je sais pas trop ce qu’on dit en chimie dans des cas pareils. Haut ou bas spin c’est pareil ?
Car un ligand est fort s’il est chargé, si on le compare à sa forme neutre. Exemple :
$$HO^- > H_2O$$
$$EtO^- > EtOH$$
Déjà, c’est un point. Ensuite les alcools de manière generale, ne complexe pas grand chose. Les Oxygènes sont mauvais (sauf dans le cas de carboxylate où ils sont moyennement bons) comparé aux soufre/azote.
Preuve, c’est le carbone qui se lie au métal dans le cas du ligand carbonyl $CO$ et non pas l’oxygène.
Connectez-vous pour pouvoir poster un message.
Connexion
Pas encore membre ?
Créez un compte en une minute pour profiter pleinement de toutes les fonctionnalités de Zeste de Savoir. Ici, tout est gratuit et sans publicité.
Créer un compte
 )
)